后胚胎环 / ARS
后胚胎环(PE):向前移位并增厚的Schwalbe线。在裂隙灯显微镜下,角膜缘内侧可见灰白色同心圆线。
阿克森费尔德异常:后胚胎环伴有周边虹膜组织索状粘连。
里格异常:除上述表现外,因虹膜基质发育不全导致瞳孔偏位、葡萄膜外翻和假性多瞳。常染色体显性遗传。50%~60%并发青光眼。

前眼部发育异常(Anterior Segment Developmental Anomalies; ASDA)是与眼前部——角膜、虹膜、晶状体、前房——相关的发育障碍的总称。也称为前眼部形成异常(Anterior Segment Dysgenesis; ASD)。
ASDA包括以下代表性病种。
这些疾病在表型和基因型上都具有多样性,已发现超过50个基因与之相关。尽管通过外显子组分析和全基因组分析,遗传学知识不断扩展,但仍有40%–75%的病例未能确定致病基因。无法归类为特定表型的病例被描述为“未分类ASD”。1)
由虹膜睫状体产生的房水通过小梁网进入施莱姆管,并经由葡萄膜巩膜流出通路排出。在ASDA中,这一过程常受损,继发性青光眼是常见且重要的并发症。
如果仅存在后胚胎环而无全身症状,则根据世界青光眼协会第9次共识报告,将其与ARS区分处理。1)
因疾病而异。原发性先天性青光眼多在出生后一年内发病。Axenfeld-Rieger综合征和Peters异常通常在出生时即被诊断。晚发型发育性青光眼可能延迟至10~20岁发病。无论哪种情况,早期发现和早期治疗都很重要。
在婴幼儿期,眼压升高引起的以下症状常作为首发症状出现。
在年龄较大的儿童和成人中,迟发型病例可能从相对年轻时就开始出现视物模糊或视力下降。当眼压非常高时,可能出现眼疲劳或头痛等症状。无虹膜症患者可能主诉畏光(昼盲)。
牛眼(角膜直径增大)和角膜混浊常由家长发现,从而促使就医。
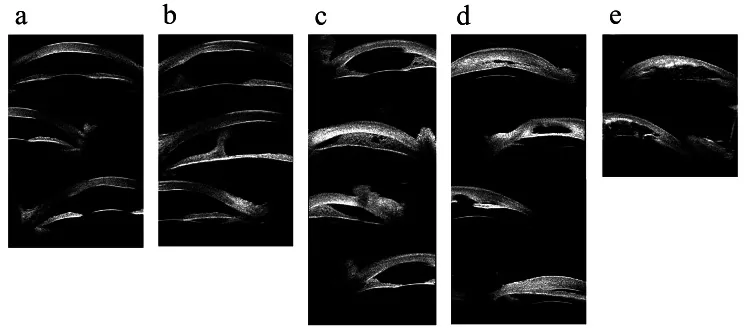
ASDA根据疾病不同呈现特征性表现。以下列出代表性疾病单元的主要所见。
后胚胎环 / ARS
后胚胎环(PE):向前移位并增厚的Schwalbe线。在裂隙灯显微镜下,角膜缘内侧可见灰白色同心圆线。
阿克森费尔德异常:后胚胎环伴有周边虹膜组织索状粘连。
里格异常:除上述表现外,因虹膜基质发育不全导致瞳孔偏位、葡萄膜外翻和假性多瞳。常染色体显性遗传。50%~60%并发青光眼。
彼得斯异常
无虹膜症
角膜异常型
合并继发性青光眼时,可能出现以下表现。
50–60%(部分报告为50–75%)的患者发生青光眼,频率很高。3) 呈常染色体显性遗传。伴有全身症状(牙齿异常、面骨异常、垂体异常等)者称为Rieger综合征。建议对亲属进行青光眼筛查。
ASDA的主要原因是遗传异常,不同疾病涉及不同的基因和遗传方式。主要疾病的致病基因如下所示。
| 疾病 | 主要致病基因 | 遗传方式 |
|---|---|---|
| ARS | PITX2(4q25)、FOXC1(6p25) | 常染色体显性遗传 |
| 彼得斯异常 | PAX6、PITX2、CYP1B1 | 散发性、显性、隐性 |
| 原发性先天性青光眼 | CYP1B1(GLC3A)、LTBP2(GLC3C) | 常染色体隐性遗传 |
| 无虹膜症 | PAX6(11号染色体) | 常染色体显性 |
| CHED | SLC4A11、ZEB1 | 常染色体隐性 |
| 巨大角膜 | CHRDL1 | X连锁隐性遗传 |
此外,还报道了伴有PAX6、PITX2、FOXC1等基因异常的发育性青光眼。基因型与表型的相关性多样,即使在同一基因异常的家族内,表型也可能不同。
早发型发育性青光眼(原发性先天性青光眼)大部分为散发病例,但约10%呈常染色体隐性遗传。也有多基因遗传的说法。
神经嵴细胞在前段形成中起核心作用。小梁网细胞来源于神经嵴,而近小管结缔组织来源于血管内皮细胞。这些起源不同的组织相邻处存在最大的房水流出阻力。ARS、彼得斯异常和先天性虹膜外翻综合征均被视为由神经嵴细胞迁移异常引起的先天异常。
韩国的一项大规模研究表明,母亲在受孕前三个月以及妊娠第一和第二孕期暴露于PM2.5(细颗粒物)增加,与后代ASDA风险升高相关。
ASDA的诊断主要基于临床。5岁及以下儿童进行检查时,通常需要在全身麻醉或镇静下进行。
根据日本青光眼学会青光眼诊疗指南第4版,满足以下2项或以上即可诊断为儿童青光眼。
与表现为角膜混浊和角膜直径增大的疾病的鉴别诊断如下所示。
ASD包含的疾病群(Axenfeld-Rieger异常、Peters异常、无虹膜症、后部多形性营养不良、小眼球、小角膜等)需要在鉴别诊断中相互考虑。3)
单独的后胚胎环(无全身症状)与ARS不同,但也是ARS的合并表现之一。后胚胎环也可能在健康眼中观察到,其本身不一定意味着青光眼风险。但若伴有其他疾病如Alagille综合征,则需要监测眼压。
ASDA相关青光眼的治疗参照原发性先天性青光眼(PCG)的治疗。
药物治疗是旨在术前短期降眼压和术后眼压控制的辅助治疗。药物选择基本与成人开角型青光眼相同。但β受体阻滞剂需注意支气管哮喘和心动过缓,新生儿有呼吸暂停的报道。也可口服乙酰唑胺(5~10 mg/kg,每6~8小时一次)。
早发型发育性青光眼基本需要手术治疗。药物治疗为辅助地位。
彼得斯异常的治疗参照PCG,但术后获得良好眼压的比例仅约手术病例的1/3,预后不良者多。由于伴有角膜异常等,常难以获得实用视力。4)
无虹膜症伴发的青光眼也参照PCG进行治疗。4)
彼得斯异常:轻症病例中角膜混浊常逐渐减轻。若眼压正常,多有改善,且因角膜移植术后预后不良,通常不在幼儿期进行角膜移植。对青光眼药物治疗抵抗,即使进行流出道重建手术也难以控制的预后不良病例较多。
CHED:针对角膜内皮功能不全,可考虑角膜移植(包括内皮移植)。
巩膜化角膜:可能与其他ASD综合征相关,重症病例可考虑角膜移植。
即使眼压下降,弱视治疗也常常是必要的。由于屈光参差、不规则散光、角膜混浊和Haab线等可导致弱视,视力及屈光检查应与眼压测量并行持续进行。近视进展和眼轴延长提示青光眼进展,因此需要定期测量。
正常前节形成遵循复杂的发育程序。妊娠第3周初,神经板形成视沟,是视觉器官发育的开始。第3周末形成视泡,第4周形成视杯。第6周左右开始闭合胚裂,第7周完成。覆盖晶状体前面的间充质分离形成前房。
神经嵴细胞从神经嵴脱上皮化,经过上皮-间充质转化迁移到眼内各处。小梁细胞来源于神经嵴,近施莱姆管结缔组织来源于血管内皮细胞,这种起源差异形成了房水流出最大阻力的部位。
ASDA中的继发性青光眼主要由于房水流出通道发育不全引起。具体涉及以下复合因素。
ICE综合征的病因与其他ASDA不同。有病毒病因学说认为单纯疱疹病毒(HSV)参与角膜内皮细胞变性,但尚未确定。该病为后天性,发生于中年成人(女性略多),通常为单眼,这也与其他ASDA不同。
无虹膜患者终生出现进行性角膜混浊。角膜缘干细胞缺乏(LSCD)被认为是主要机制。多项确认PAX6突变的研究记录了这种进行性变化。发病率报道为20-80%以上,常对称出现但不总是如此。2)
通过外显子组分析和全基因组分析,新的相关基因的鉴定正在推进。然而,仍有40-75%的病例未能确定致病基因,对剩余的“未解病例”进行分析是未来的重要课题。阐明基因型与临床表型之间的关联有望应用于个体化医疗。
在携带FOXC1和PITX2突变的病例中,青光眼的发病年龄和临床表现存在差异。虽然基因型可能与表型多样性相关,但同一基因突变也可能呈现不同的疾病类型,这使得诊断和预后预测变得困难。1)
无虹膜症相关角膜病变(AAK)的发病率据报道为20%至80%以上,多项经PAX6突变确认的研究记录了终生角膜混浊的进展。针对LCSD的角膜缘干细胞移植研究正在推进,但目前仍处于研究阶段,尚未确立为标准治疗。2)
流行病学研究表明,受精前和妊娠期间暴露于大气污染(PM2.5)与ASDA风险相关,从环境预防医学的角度正在探索公共卫生应用。这可能为未来的预防策略提供方向。
微脉冲激光和微创青光眼手术(MIGS)设备在ASDA儿童中的应用处于研究阶段。长期结果数据有限,尚未确立与成人青光眼同等的有效性和安全性。