Las anomalías del desarrollo del segmento anterior (Anterior Segment Developmental Anomalies; ASDA) son un término general para los trastornos del desarrollo relacionados con el segmento anterior del ojo: córnea, iris, cristalino y cámara anterior. También se denomina disgenesia del segmento anterior (Anterior Segment Dysgenesis; ASD).
ASDA incluye las siguientes entidades patológicas representativas.
Estas enfermedades son diversas tanto en fenotipo como en genotipo, y se ha descubierto que más de 50 genes están involucrados. Aunque el conocimiento genético continúa expandiéndose mediante el análisis de exomas y el análisis del genoma completo, el gen causante aún no se ha identificado en el 40–75% de los casos. Los casos que no pueden clasificarse en un fenotipo específico se describen como “ASD no clasificado”. 1)
El humor acuoso producido por el cuerpo ciliar del iris se drena a través de la malla trabecular hacia el canal de Schlemm y a través de la vía de salida uveoescleral. En la ASDA, este proceso suele verse afectado, y el glaucoma secundario es una complicación común e importante.
Si solo está presente el embriotoxón posterior sin síntomas sistémicos, se trata por separado del ARS según el 9.º Informe de Consenso de la Asociación Mundial de Glaucoma. 1)
Q¿A qué edad se diagnostica típicamente la anomalía del desarrollo del segmento anterior (ASDA)?
A
Varía según la enfermedad. El glaucoma congénito primario a menudo se desarrolla dentro del primer año de vida. El síndrome de Axenfeld-Rieger y la anomalía de Peters a menudo se diagnostican al nacer. El glaucoma de desarrollo de inicio tardío puede retrasarse hasta la adolescencia o los veinte años. La detección y el tratamiento tempranos son importantes en todos los casos.
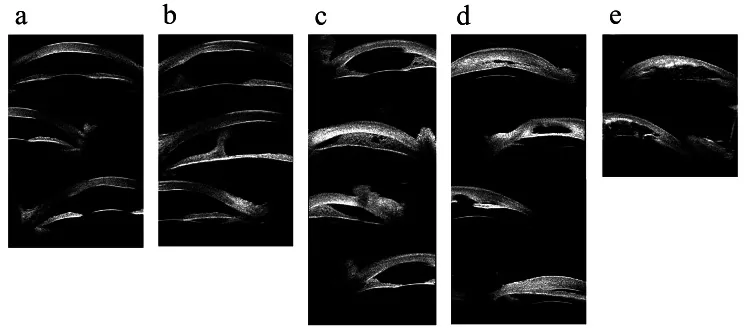
Hong J, et al. Classifications of anterior segment structure of congenital corneal opacity in infants and toddlers by ultrasound biomicroscopy and slit-lamp microscopic photographs: an observational study. BMC Ophthalmol. 2024. Figure 1. PMCID: PMC10804776. License: CC BY.
(a) Opacidad corneal, (b) Opacidad corneal con sinequia anterior central, (c) Adherencia iridocorneal periférica ≤180 grados, (d) Adherencia iridocorneal periférica >180 grados, (e) Imagen de biomicroscopía ultrasónica de opacidad corneal con anomalías del iris y cristalino. Corresponden a las adherencias del segmento anterior y opacidades corneales tratadas en la sección “2. Principales síntomas y hallazgos clínicos”.
En la infancia, los siguientes síntomas debidos al aumento de la presión intraocular suelen aparecer como síntomas iniciales.
Lagrimeo (epífora): Ocurre como resultado de la irritación por edema del epitelio corneal debido al aumento de la presión intraocular.
Fotofobia (sensibilidad a la luz): Síntoma que refleja la irritación corneal.
Blefaroespasmo: Aparece por el mismo mecanismo que el lagrimeo y la fotofobia.
En niños mayores y adultos, los casos de inicio tardío pueden presentar visión borrosa o disminución de la agudeza visual desde una edad relativamente temprana. Cuando la presión intraocular es muy alta, pueden aparecer síntomas como fatiga ocular o dolor de cabeza. En la aniridia, los pacientes pueden quejarse de fotofobia (ceguera diurna).
El buftalmos (aumento del diámetro corneal) y la opacidad corneal suelen ser descubiertos por los padres, lo que motiva la consulta médica.
La ASDA presenta hallazgos característicos según la enfermedad. A continuación se muestran los principales hallazgos de las entidades nosológicas representativas.
Embriotóxon posterior / ARS
Embriotóxon posterior (PE): Línea de Schwalbe desplazada anteriormente y engrosada. Se observa como una línea concéntrica de color gris blanquecino en la parte interna del limbo corneal bajo microscopía de lámpara de hendidura.
Anomalía de Axenfeld: Embriotóxon posterior con adherencias en forma de cordón del tejido iridiano periférico.
Anomalía de Rieger: Además de lo anterior, presenta corectopia, ectropión uveal y pseudopolicoria debido a hipoplasia del estroma del iris. Herencia autosómica dominante. El 50–60% desarrolla glaucoma.
Anomalía de Peters
Opacidad corneal central: Hallazgo esencial para el diagnóstico. Refleja defectos en el endotelio corneal, la membrana de Descemet y el estroma corneal.
Tipo 1: Solo defecto corneal posterior y opacidad corneal.
Tipo 2: Acompañado de adherencias del iris.
Tipo 3: Acompañado de desplazamiento anterior del cristalino y cataratas. Aproximadamente el 80% son bilaterales. Se asocia con glaucoma en el 50–70% de los casos.
Aniridia
Hipoplasia del iris: Principalmente un defecto de la porción posterior del iris. Puede acompañarse de hipoplasia macular, hipoplasia del nervio óptico y glaucoma.
Queratopatía asociada a aniridia (AAK): La incidencia reportada varía del 20% a más del 80%. Es una opacidad corneal progresiva debida a insuficiencia de células madre del limbo (LSCD) que progresa a lo largo de la vida. 2)
Síndrome de WAGR: Ocurre cuando PAX6 y el gen WT1 adyacente mutan. Incluye tumor de Wilms, aniridia, anomalías genitourinarias y discapacidad intelectual. 3)
Tipo de anomalía corneal
Megalocórnea: Diámetro corneal ≥13 mm (≥12 mm en recién nacidos). Generalmente la presión intraocular y la densidad de células endoteliales son normales. Con frecuencia herencia recesiva ligada al cromosoma X.
Esclerocórnea: Tejido escleral opaco invade la córnea periférica. El borde entre esclerótica y córnea es indistinto y se acompaña de invasión vascular.
CHED: Edema corneal bilateral simétrico que aparece al nacer hasta los 1-2 años de edad. No se asocia con elevación de la presión intraocular. Autosómico recesivo.
A continuación se muestran los hallazgos que pueden agregarse cuando se complica con glaucoma secundario.
Elevación de la presión intraocular: Puede presentar presión intraocular alta (aproximadamente 30-50 mmHg).
Aumento del diámetro corneal (buftalmos): Debido a la distensión de la cubierta ocular. Si el diámetro supera los 12.0 mm inmediatamente después del nacimiento, se debe sospechar glaucoma congénito.
Estrías de Haab: Opacidades lineales permanentes que quedan en el sitio de la ruptura de la membrana de Descemet.
Agrandamiento de la excavación del disco óptico: En lactantes, una relación copa-disco de 0.3 o más sugiere glaucoma. Una diferencia de 0.2 o más entre ambos ojos también es sospechosa.
Q¿Qué porcentaje de pacientes con síndrome de Axenfeld-Rieger desarrolla glaucoma?
A
Se desarrolla glaucoma en el 50–60% (algunos informes 50–75%) de los casos, una frecuencia alta. 3) Sigue un patrón de herencia autosómico dominante. Los casos con hallazgos sistémicos (anomalías dentales, anomalías de los huesos faciales, anomalías hipofisarias, etc.) se denominan síndrome de Rieger. Se recomienda el cribado de glaucoma en los familiares.
La causa principal de ASDA son las anomalías genéticas, con diferentes genes y patrones de herencia según la enfermedad. Los genes causantes de las principales enfermedades se muestran a continuación.
Además, se ha informado de glaucoma del desarrollo asociado con anomalías genéticas como PAX6, PITX2 y FOXC1. La correlación entre genotipo y fenotipo es diversa, y el fenotipo puede diferir incluso dentro de familias con la misma anomalía genética.
La mayoría de los casos de glaucoma del desarrollo de inicio temprano (glaucoma congénito primario) son esporádicos, pero aproximadamente el 10% sigue un patrón de herencia autosómico recesivo. También existe una teoría de herencia multifactorial.
Las células de la cresta neural desempeñan un papel central en la formación del segmento anterior. Las células de la malla trabecular derivan de la cresta neural, mientras que el tejido conectivo yuxtacanalicular deriva de células endoteliales vasculares. La mayor resistencia al flujo de humor acuoso existe en la unión de estos tejidos de diferentes orígenes. El ARS, la anomalía de Peters y el síndrome de ectropión uveal congénito se clasifican como anomalías congénitas resultantes de una migración anormal de las células de la cresta neural.
Un estudio a gran escala en Corea mostró que un aumento en la exposición materna a PM2.5 (material particulado fino) durante los tres meses previos a la concepción y durante el primer y segundo trimestre del embarazo se asoció con un mayor riesgo de ASDA en la descendencia.
Examen con lámpara de hendidura: Evalúa el grado y la ubicación de la opacidad corneal, presencia de estrías de Haab, profundidad de la cámara anterior, anomalías del iris (embriotóxon posterior, inserción del iris en la línea de Schwalbe) y anomalías del cristalino. Verifica la presencia de embriotóxon posterior, anomalías del iris (ARS) y cataratas (anomalía de Peters).
Medición de la presión intraocular: La tonometría de aplanación de Goldmann es estándar, pero en niños son útiles los tonómetros portátiles como el tonómetro de rebote (iCare) o el tonómetro electrónico (Tono-Pen). Tenga en cuenta que la presión intraocular disminuye bajo anestesia general. Las mediciones no son intercambiables entre diferentes tonómetros.
Medición del diámetro corneal: Mide los diámetros horizontal y vertical con un calibrador. El rango normal en recién nacidos es de 9.5 a 10.5 mm. Si supera los 12.0 mm inmediatamente después del nacimiento, se sospecha glaucoma congénito.
Gonioscopia: Utiliza una lámpara de hendidura portátil y un gonioscopio directo como la lente de Koeppe. Evalúa la inserción alta del iris, la inserción del iris en la línea de Schwalbe (hallazgo de ARS) y el aumento del ancho de la malla trabecular.
Examen de fondo de ojo: Observación de la excavación del disco óptico. En lactantes, una relación copa-disco ≥0.3 sugiere glaucoma. La reducción de la excavación tras disminuir la presión intraocular indica buen control de la presión.
Microscopía ultrasónica biomicroscópica (UBM): Útil cuando la opacidad corneal dificulta la visualización del ángulo. Ayuda a evaluar el grado de disgenesia angular y a predecir el pronóstico de la cirugía de reconstrucción de la vía de salida.
OCT de segmento anterior (AS-OCT): Como prueba complementaria, evalúa de forma no invasiva la estructura del ángulo y la córnea, pero no reemplaza a la gonioscopia para el diagnóstico. 3)
Campimetría: Esencial para el diagnóstico de la neuropatía óptica glaucomatosa infantil. En niños menores de 5 años, es difícil incluso para examinadores experimentados; la perimetría cinética es más fácil de realizar.
Según la 4ª edición de las Guías de Práctica Clínica para Glaucoma de la Sociedad Japonesa de Glaucoma, se diagnostica glaucoma infantil cuando se cumplen dos o más de los siguientes criterios.
Presión intraocular > 21 mmHg
Progresión del aumento de la relación copa-disco, asimetría de la relación copa-disco ≥ 0.2, adelgazamiento del borde
Hallazgos corneales: estrías de Haab, o en recién nacidos diámetro corneal ≥ 11 mm, en menores de 1 año ≥ 12 mm, en todas las edades ≥ 13 mm
Progresión de la miopía por alargamiento axial más allá del desarrollo normal
Defecto de campo visual reproducible consistente con neuropatía óptica glaucomatosa
Traumatismo por fórceps: Unilateral, opacidades lineales verticales u oblicuas.
Enfermedades metabólicas congénitas como mucopolisacaridosis y cistinuria: La evaluación de los síntomas sistémicos es importante para el diagnóstico diferencial.
Los grupos de enfermedades incluidos en ASD (anomalía de Axenfeld-Rieger, anomalía de Peters, aniridia, distrofia polimorfa posterior, microftalmia, microcórnea, etc.) deben considerarse mutuamente en el diagnóstico diferencial. 3)
Q¿El embryotoxon posterior aislado también se asocia con glaucoma?
A
El embryotoxon posterior aislado (sin síntomas sistémicos) se distingue del ARS, pero también es uno de los hallazgos asociados del ARS. El embryotoxon posterior también se puede observar en ojos sanos y no necesariamente indica un riesgo de glaucoma. Sin embargo, cuando se asocia con otras enfermedades como el síndrome de Alagille, es necesario el seguimiento de la presión intraocular.
La farmacoterapia es un tratamiento adyuvante destinado a la reducción de la presión intraocular a corto plazo antes de la cirugía y al control de la presión postoperatoria. La selección de fármacos es generalmente la misma que para el glaucoma de ángulo abierto del adulto. Sin embargo, los betabloqueantes requieren precaución por asma bronquial y bradicardia, y se ha informado apnea en neonatos. También es posible la administración oral de acetazolamida (5–10 mg/kg cada 6–8 horas).
El glaucoma de desarrollo de inicio temprano es una enfermedad que fundamentalmente requiere terapia quirúrgica. La farmacoterapia es adyuvante.
Goniotomía: Adecuada como cirugía inicial en casos con opacidad corneal mínima. Tiene la ventaja de no invadir la conjuntiva. Usando una lente de Barkan o Swan-Jacob, se raspa la superficie de la malla trabecular con un cuchillo de goniotomía.
Trabeculotomía: Se puede realizar independientemente de la opacidad corneal. También se realiza como cirugía adicional cuando la goniotomía es insuficientemente efectiva.
Trabeculectomía/Cirugía de derivación con tubo: Opciones cuando la cirugía del ángulo es ineficaz. En ARS, se elige la cirugía del ángulo si el ángulo está abierto y la cobertura de la malla trabecular por sinequias anteriores periféricas no es extensa, pero la tasa de éxito es menor que en PCG. En casos ineficaces de cirugía del ángulo, la trabeculectomía o la cirugía de derivación con tubo y placa pueden ser la primera opción. 4)
En la anomalía de Peters, el tratamiento sigue el mismo enfoque que para PCG, pero se logra una buena presión intraocular postoperatoria solo en aproximadamente un tercio de los casos quirúrgicos, y muchos tienen mal pronóstico. Debido a las anomalías corneales, a menudo es difícil lograr una visión útil. 4)
El glaucoma asociado con aniridia también se trata siguiendo el mismo enfoque que para PCG. 4)
Anomalía de Peters: En casos leves, la opacidad corneal a menudo disminuye gradualmente. Si la presión intraocular es normal, suele haber cierta mejoría, y debido al mal resultado del trasplante de córnea, generalmente no se realiza en la primera infancia. Muchos casos son refractarios al tratamiento farmacológico del glaucoma y a la cirugía de reconstrucción de la vía de salida, con mal pronóstico.
CHED: El trasplante de córnea (incluido el trasplante endotelial) puede estar indicado para la disfunción endotelial corneal.
Esclerocórnea: Puede asociarse con otros síndromes ASD; los casos graves pueden ser candidatos a trasplante de córnea.
Incluso si la presión intraocular disminuye, a menudo es necesario el tratamiento de la ambliopía. Dado que la anisometropía, el astigmatismo irregular, la opacidad corneal y las estrías de Haab pueden causar ambliopía, los exámenes de agudeza visual y refracción deben continuarse en paralelo con la medición de la presión intraocular. La progresión de la miopía y el alargamiento de la longitud axial sugieren progresión del glaucoma, por lo que se requieren mediciones periódicas.
La formación normal del segmento anterior sigue un programa de desarrollo complejo. Al comienzo de la tercera semana de gestación, se forma el surco óptico en la placa neural, marcando el inicio del desarrollo del órgano visual. Al final de la tercera semana se forma la vesícula óptica, y en la cuarta semana se forma la copa óptica. El cierre de la fisura embrionaria comienza alrededor de la sexta semana y se completa en la séptima semana. El mesénquima que cubre la superficie anterior del cristalino se separa para formar la cámara anterior.
Las células de la cresta neural se desepitelizan de la cresta neural y migran a varias ubicaciones dentro del ojo a través de la transición epitelio-mesénquima. Las células de la malla trabecular derivan de la cresta neural, mientras que el tejido conectivo yuxtacanalicular deriva de células endoteliales vasculares. Esta diferencia de origen forma el sitio de mayor resistencia al flujo de salida del humor acuoso.
PITX2: Factor de transcripción. Cromosoma 4 (4q25). Asociado tanto con síntomas oculares como auditivos en el síndrome de Axenfeld-Rieger.
FOXC1: Factor de transcripción. Cromosoma 6 (6p25). Implicado en el síndrome de Axenfeld-Rieger y asociado tanto con síntomas oculares como auditivos, similar a PITX2.
El glaucoma secundario en ASDA se produce principalmente por un desarrollo deficiente de la vía de drenaje del humor acuoso. Específicamente, intervienen los siguientes factores de forma combinada.
Desarrollo inmaduro de la malla trabecular: El tejido conectivo yuxtacanalicular es anormalmente grueso, con acumulación excesiva de matriz extracelular.
Inserción del cuerpo ciliar en la malla trabecular: La contracción del músculo ciliar tira del espolón escleral hacia adelante, comprimiendo el canal de Schlemm y la malla trabecular.
Inserción alta de la raíz del iris: la raíz del iris se encuentra en la posición de la malla trabecular, obstruyendo la salida del humor acuoso.
Hipoplasia o ausencia del canal de Schlemm.
El síndrome ICE tiene una etiología diferente a otras ASDA. Se ha propuesto una etiología viral que involucra al virus del herpes simple (VHS) en la degeneración de las células endoteliales corneales, pero no se ha confirmado. Es adquirido, ocurre en adultos de mediana edad (ligeramente más común en mujeres) y generalmente es unilateral, lo que también difiere de otras ASDA.
Fisiopatología de la Queratopatía Asociada a Aniridia (AAK)
Los pacientes con aniridia desarrollan opacificación corneal progresiva a lo largo de la vida. La deficiencia de células madre del limbo corneal (LSCD) se considera el mecanismo principal. Múltiples estudios con mutaciones confirmadas de PAX6 han documentado este cambio progresivo. La incidencia se reporta entre 20 y 80% o más, y a menudo aparece simétricamente pero no siempre. 2)
7. Investigación más reciente y perspectivas futuras
El análisis del exoma y el análisis del genoma completo están avanzando en la identificación de nuevos genes asociados. Sin embargo, el gen causante sigue sin identificarse en el 40-75% de los casos, y el análisis de los “casos no explicados” restantes es un desafío importante para el futuro. Se espera que la elucidación de la correlación entre el genotipo y el fenotipo clínico conduzca a aplicaciones en la medicina personalizada.
En casos con mutaciones de FOXC1 y PITX2, la edad de inicio y las características clínicas del glaucoma varían. Si bien el genotipo puede estar asociado con la diversidad fenotípica, incluso la misma mutación genética puede presentar diferentes tipos de enfermedad, lo que dificulta el diagnóstico y la predicción del pronóstico. 1)
La incidencia de la queratopatía asociada a aniridia (AAK) se reporta entre el 20 y el 80% o más, y múltiples estudios con mutaciones confirmadas de PAX6 han documentado la progresión de la opacificación corneal a lo largo de la vida. La investigación sobre el trasplante de células madre del limbo corneal dirigido a LCSD está avanzando, pero aún se encuentra en fase de investigación y no se ha establecido como tratamiento estándar. 2)
Estudios epidemiológicos han mostrado una asociación entre la exposición a la contaminación del aire (PM2.5) antes de la fertilización y durante el embarazo y el riesgo de ASDA, y se están explorando aplicaciones de salud pública desde la perspectiva de la medicina preventiva ambiental. Esto podría conducir a futuras estrategias de prevención.
Aplicación de Dispositivos Quirúrgicos Mínimamente Invasivos
La aplicación de láseres de micropulso y dispositivos de cirugía de glaucoma mínimamente invasiva (MIGS) en niños con ASDA se encuentra en fase de investigación. Los datos de resultados a largo plazo son limitados y no se ha establecido una eficacia y seguridad equivalentes a las del glaucoma en adultos.
Knight LSW, Ruddle JB, Taranath DA, et al. Childhood and Early Onset Glaucoma Classification and Genetic Profile in a Large Australasian Disease Registry. Ophthalmology. 2021;128(11):1549-1560. doi:10.1016/j.ophtha.2021.04.016.
Hu JCW, Trief D. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:13. doi:10.21037/aes-22-35. https://aes.amegroups.org/article/view/7385/html
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.