Anomalien der vorderen Augenabschnittsentwicklung (Anterior Segment Developmental Anomalies; ASDA) sind ein Sammelbegriff für Entwicklungsstörungen des vorderen Augenabschnitts – Hornhaut (Cornea), Regenbogenhaut (Iris), Linse (Lens) und vordere Augenkammer (Anterior Chamber). Sie werden auch als vordere Augenabschnittsdysgenesie (Anterior Segment Dysgenesis; ASD) bezeichnet.
ASDA umfasst die folgenden repräsentativen Krankheitseinheiten.
Diese Erkrankungen sind sowohl phänotypisch als auch genotypisch vielfältig, und es wurde festgestellt, dass über 50 Gene beteiligt sind. Exom- und Genomanalysen erweitern ständig die genetischen Erkenntnisse, dennoch wird bei 40–75 % der Fälle das ursächliche Gen nicht identifiziert. Fälle, die keinem spezifischen Phänotyp zugeordnet werden können, werden als „nicht klassifizierte ASD“ bezeichnet. 1)
Das vom Ziliarkörper der Iris produzierte Kammerwasser wird über das Trabekelwerk in den Schlemm-Kanal und über den uveoskleralen Abflussweg abgeleitet. Bei ASD ist dieser Prozess häufig gestört, und ein sekundäres Glaukom ist eine häufige und wichtige Komplikation.
Wenn nur der hintere Embryonalring ohne systemische Symptome vorliegt, wird er gemäß dem 9. Konsensbericht der World Glaucoma Association getrennt von ARS behandelt. 1)
QIn welchem Alter wird eine Anomalie der Vorderabschnittsentwicklung (ASDA) typischerweise diagnostiziert?
A
Die Symptome variieren je nach Erkrankung. Das primäre angeborene Glaukom tritt häufig im ersten Lebensjahr auf. Das Axenfeld-Rieger-Syndrom und die Peters-Anomalie werden oft bei der Geburt diagnostiziert. Das spät einsetzende Entwicklungsglaukom kann sich bis zum Alter von 10 bis 20 Jahren verzögern. In allen Fällen sind Früherkennung und frühzeitige Behandlung wichtig.
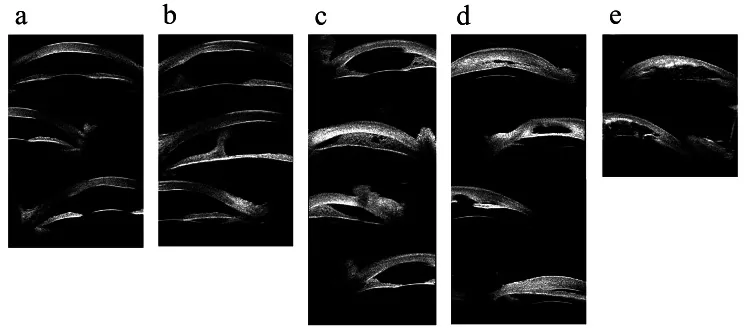
Hong J, et al. Classifications of anterior segment structure of congenital corneal opacity in infants and toddlers by ultrasound biomicroscopy and slit-lamp microscopic photographs: an observational study. BMC Ophthalmol. 2024. Figure 1. PMCID: PMC10804776. License: CC BY.
(a) Hornhauttrübung, (b) Hornhauttrübung mit zentraler vorderer Synechie, (c) periphere Iridokornealverwachsung ≤180°, (d) periphere Iridokornealverwachsung >180°, (e) Ultraschallbiomikroskopiebild einer Hornhauttrübung mit Iris- und Linsenanomalien. Entspricht den im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelten vorderen Synechien und Hornhauttrübungen.
Im Säuglingsalter werden die folgenden Symptome im Zusammenhang mit erhöhtem Augeninnendruck häufig als Erstsymptome beobachtet.
Tränenfluss (Epiphora) : Entsteht als Folge der Reizung durch ein Hornhautepithelödem aufgrund erhöhten Augeninnendrucks.
Photophobie (Lichtempfindlichkeit) : Symptom, das eine Hornhautreizung widerspiegelt.
Blepharospasmus (Lidkrampf) : Tritt durch denselben Mechanismus wie Tränenfluss und Photophobie auf.
Bei älteren Kindern und Erwachsenen kann bei der späten Form bereits in relativ jungen Jahren verschwommenes Sehen oder eine verminderte Sehschärfe auftreten. Bei sehr hohem Augeninnendruck können Symptome wie Augenermüdung oder Kopfschmerzen auftreten. Bei Aniridie kann eine Lichtempfindlichkeit (Tagblindheit) beklagt werden.
Buphthalmus (Vergrößerung des Hornhautdurchmessers) und Hornhauttrübung werden oft von den Eltern entdeckt und führen zur Konsultation.
Die ASDA zeigt je nach Erkrankung charakteristische Befunde. Die wichtigsten Befunde repräsentativer Krankheitseinheiten sind unten aufgeführt.
Posteriorer Embryotoxon / ARS
Posteriorer Embryotoxon (PE) : Nach vorne verlagerte und verdickte Schwalbe-Linie. Im Spaltlampenmikroskop als grau-weiße konzentrische Linie innerhalb des Hornhautlimbus sichtbar.
Axenfeld-Anomalie : hinterer Embryotoxon mit strangförmigen Verwachsungen des peripheren Irisgewebes.
Rieger-Anomalie: Zusätzlich zu den oben genannten Befunden zeigen sich Pupillenverlagerung, Uveaektropium und Pseudopolykorie aufgrund einer Irisstromahypoplasie. Autosomal-dominanter Erbgang. In 50–60 % der Fälle tritt ein Glaukom auf.
Peters-Anomalie
Zentrale Hornhauttrübung : für die Diagnose obligater Befund. Spiegelt Defekte des Hornhautendothels, der Descemet-Membran und des Hornhautstromas wider.
Typ 1 : nur hinterer Hornhautdefekt und Hornhauttrübung.
Typ 2 : Mit Irisverwachsungen.
Typ 3 : Linsenverschiebung nach vorne oder Katarakt. Etwa 80% sind beidseitig. Glaukom tritt in 50–70% der Fälle auf.
Aniridie
Iris-Hypoplasie : Hauptsächlich Defekt der hinteren Iris. Kann mit Makulahypoplasie, Optikushypoplasie oder Glaukom einhergehen.
Aniridie-assoziierte Keratopathie (AAK) : Die Inzidenz wird mit 20–80% oder höher angegeben. Progressive Hornhauttrübung durch Limbusstammzellinsuffizienz (LSCD), die lebenslang fortschreitet. 2)
WAGR-Syndrom : Tritt bei Mutation des PAX6- und des benachbarten WT1-Gens auf. Umfasst Wilms-Tumor, Aniridie, urogenitale Anomalien und geistige Entwicklungsverzögerung. 3)
Hornhautanomalie-Typ
Megalokornea : Hornhautdurchmesser ≥13 mm (bei Neugeborenen ≥12 mm). Normalerweise normaler Augeninnendruck und Endothelzelldichte. Häufig X-chromosomal rezessiv vererbt.
Skleralisierte Hornhaut : undurchsichtiges Skleragewebe dringt in die periphere Hornhaut ein. Hornhaut-Sklera-Grenze unscharf mit Gefäßeinsprossung.
CHED : beidseitiges symmetrisches Hornhautödem bei Geburt bis zum Alter von 1-2 Jahren. Kein erhöhter Augeninnendruck. Autosomal rezessiv.
Bei begleitendem sekundärem Glaukom kommen folgende Befunde hinzu.
Erhöhter Augeninnendruck : kann hohen Augeninnendruck (ca. 30-50 mmHg) aufweisen.
Vergrößerter Hornhautdurchmesser (Buphthalmus) : durch Dehnung der Augenhülle. Bei einem Durchmesser >12,0 mm direkt nach der Geburt ist ein kongenitales Glaukom zu vermuten.
Haab-Linie : dauerhafte lineare Trübung an der Stelle des Risses der Descemet-Membran.
Vergrößerung der Papillenexkavation : Bei Säuglingen und Kleinkindern lässt ein C/D-Verhältnis von über 0,3 auf ein Glaukom schließen. Ein Seitenunterschied von 0,2 oder mehr ist ebenfalls verdächtig.
QBei wie viel Prozent der Patienten mit Axenfeld-Rieger-Syndrom entwickelt sich ein Glaukom?
A
Bei 50–60 % (einige Berichte geben 50–75 % an) tritt ein Glaukom auf, was eine hohe Häufigkeit darstellt. 3) Es liegt ein autosomal-dominanter Erbgang vor. Fälle mit systemischen Symptomen (Zahnanomalien, Gesichtsknochenanomalien, Hypophysenanomalien usw.) werden als Rieger-Syndrom bezeichnet. Ein Glaukom-Screening bei Verwandten wird empfohlen.
Die Hauptursache der ASDA sind genetische Anomalien, wobei je nach Erkrankung unterschiedliche Gene und Vererbungsmuster beteiligt sind. Die ursächlichen Gene der wichtigsten Erkrankungen sind unten aufgeführt.
Darüber hinaus wurde auch über entwicklungsbedingtes Glaukom in Verbindung mit Genanomalien wie PAX6, PITX2 und FOXC1 berichtet. Die Korrelation zwischen Genotyp und Phänotyp ist vielfältig, und der Phänotyp kann innerhalb derselben Familie mit derselben Genanomalie variieren.
Die meisten Fälle von frühmanifestem entwicklungsbedingtem Glaukom (primäres kongenitales Glaukom) sind sporadisch, aber etwa 10 % folgen einem autosomal-rezessiven Erbgang. Es gibt auch die Theorie eines multifaktoriellen Erbgangs.
Neuralleistenzellen (neural crest cells) spielen eine zentrale Rolle bei der Bildung des vorderen Augenabschnitts. Die Trabekelzellen stammen von der Neuralleiste ab, während das juxtakanalikuläre Bindegewebe von vaskulären Endothelzellen abstammt. Der größte Widerstand gegen den Kammerwasserabfluss befindet sich an der Kontaktstelle dieser Gewebe unterschiedlichen Ursprungs. ARS, Peters-Anomalie und das kongenitale Iris-Eversionssyndrom werden alle als angeborene Anomalien aufgrund einer gestörten Migration von Neuralleistenzellen angesehen.
Eine groß angelegte Studie in Südkorea zeigte, dass eine erhöhte mütterliche Exposition gegenüber PM2,5 (Feinstaub) in den drei Monaten vor der Empfängnis sowie im ersten und zweiten Trimester der Schwangerschaft mit einem erhöhten Risiko für ASDA beim Kind verbunden war.
Die Diagnose der ASDA erfolgt hauptsächlich klinisch. Bei Kindern unter 5 Jahren sind Untersuchungen oft unter Vollnarkose oder Sedierung erforderlich.
Spaltlampenmikroskopie : Beurteilung des Ausmaßes und der Lokalisation der Hornhauttrübung, Vorhandensein von Haab-Linien, Tiefe der Vorderkammer, Irisanomalien (posteriorer Embryotoxon, Irisansatz an der Schwalbe-Linie) und Linsenanomalien. Überprüfung auf posterioren Embryotoxon, Irisanomalien (ARS) und Katarakt (Peters-Anomalie).
Augeninnendruckmessung : Das Goldmann-Applanationstonometer ist der Standard, aber bei Kindern sind tragbare Tonometer wie das Rebound-Tonometer (iCare) oder das elektronische Tonometer (Tono-Pen) nützlich. Beachten Sie, dass der Augeninnendruck unter Vollnarkose sinkt. Die Messwerte verschiedener Tonometer sind nicht austauschbar.
Hornhautdurchmessermessung : Messung des horizontalen und vertikalen Durchmessers mit einem Messschieber. Der normale Bereich bei Neugeborenen beträgt 9,5–10,5 mm. Ein Durchmesser über 12,0 mm unmittelbar nach der Geburt lässt an ein angeborenes Glaukom denken.
Kammerwinkeluntersuchung : Verwendung einer Handspaltlampe und eines direkten Gonioskops wie der Koeppe-Linse. Beurteilung des hohen Irisansatzes, des Irisansatzes an der Schwalbe-Linie (ARS-Zeichen) und der Verbreiterung des Trabekelwerks.
Fundusuntersuchung: Beobachtung der Papillenexkavation. Bei Säuglingen wird bei einem C/D-Verhältnis ≥ 0,3 ein Glaukom vermutet. Eine Verkleinerung der Exkavation nach Augeninnendrucksenkung ist ein Zeichen für eine gute Druckkontrolle.
Ultraschallbiomikroskopie (UBM): Nützlich bei Hornhauttrübung, die die Beurteilung des Kammerwinkels erschwert. Hilft bei der Beurteilung des Schweregrads von Kammerwinkeldysgenesie und der Prognose von Abflussrekonstruktionsoperationen.
Vorderabschnitts-OCT (AS-OCT): Kann als ergänzende Untersuchung die Struktur von Kammerwinkel und Hornhaut nicht-invasiv beurteilen, ersetzt jedoch nicht die Gonioskopie zur Diagnose. 3)
Gesichtsfelduntersuchung: Essenziell für die Diagnose einer glaukomatösen Optikusneuropathie bei Kindern. Unter 5 Jahren ist sie selbst für erfahrene Untersucher schwierig; die kinetische Perimetrie ist einfacher durchzuführen.
Laut der 4. Auflage der Glaukom-Leitlinien der Japanischen Glaukomgesellschaft wird ein kindliches Glaukom diagnostiziert, wenn mindestens zwei der folgenden Kriterien erfüllt sind.
Augeninnendruck > 21 mmHg
Progression der C/D-Ratio-Erhöhung, C/D-Ratio-Asymmetrie ≥ 0,2, Ausdünnung des Randsaums
Hornhautbefunde: Haab-Linien, oder bei Neugeborenen Hornhautdurchmesser ≥ 11 mm, bei Kindern unter 1 Jahr ≥ 12 mm, in allen Altersgruppen ≥ 13 mm
Fortschreiten der Myopie durch Verlängerung der Achsenlänge über die normale Entwicklung hinaus
Reproduzierbarer Gesichtsfeldausfall, der mit einer glaukomatösen Optikusneuropathie übereinstimmt
CHED: Beidseitiges symmetrisches Hornhautödem. Kein erhöhter Augeninnendruck.
Posteriore polymorphe Hornhautdystrophie: Keine Hornhautdurchmesservergrößerung. Hornhautendotheluntersuchung diagnostisch hilfreich.
Forceps-Geburtstrauma: Einseitig, vertikale oder schräge lineare Trübungen.
Angeborene Mukopolysaccharidose, Zystinurie und andere Stoffwechselerkrankungen: Die Beurteilung systemischer Symptome ist für die Differentialdiagnose wichtig.
Die Krankheitsgruppen, die zum ASD gehören (Axenfeld-Rieger-Anomalie, Peters-Anomalie, Aniridie, posteriore polymorphe Dystrophie, Mikrophthalmie, Mikrokornea usw.), müssen in der Differentialdiagnose gegenseitig berücksichtigt werden. 3)
QIst ein isolierter posteriorer Embryotoxon mit einem Glaukom assoziiert?
A
Ein isolierter posteriorer Embryotoxon (ohne systemische Symptome) wird von ARS unterschieden, kann aber ein Begleitbefund von ARS sein. Ein posteriorer Embryotoxon kann auch bei gesunden Augen beobachtet werden und bedeutet an sich nicht unbedingt ein Glaukomrisiko. Wenn er jedoch mit anderen Erkrankungen wie dem Alagille-Syndrom einhergeht, ist eine Überwachung des Augeninnendrucks erforderlich.
Die Behandlung des Glaukoms im Zusammenhang mit ASDA erfolgt analog zur Behandlung des primären kongenitalen Glaukoms (PCG).
Medikamentöse Therapie ist eine adjuvante Behandlung zur kurzfristigen Augeninnendrucksenkung vor der Operation und zur postoperativen Druckkontrolle. Die Medikamentenauswahl erfolgt grundsätzlich wie beim adulten Offenwinkelglaukom. Bei Betablockern ist jedoch Vorsicht bei Asthma bronchiale und Bradykardie geboten; bei Neugeborenen wurde über Apnoe berichtet. Acetazolamid oral (5–10 mg/kg alle 6–8 Stunden) ist ebenfalls möglich.
Das frühkindliche Entwicklungsglaukom erfordert grundsätzlich eine chirurgische Therapie. Die medikamentöse Therapie hat eine unterstützende Rolle.
Goniotomie : Geeignet als Ersteingriff bei Fällen mit geringer Hornhauttrübung. Vorteil: keine Verletzung der Bindehaut. Verwendung einer Barkan- oder Swan-Jacob-Linse, Abtragen der Trabekeloberfläche mit einem Goniotomiemesser.
Trabekulotomie : Unabhängig vom Vorhandensein einer Hornhauttrübung durchführbar. Wird auch als Zusatzoperation bei unzureichender Wirkung der Goniotomie durchgeführt.
Trabekulektomie und Tubus-Shunt-Operation : Optionen bei unwirksamer Kammerwinkelchirurgie. Bei ARS wird die Kammerwinkelchirurgie gewählt, wenn der Kammerwinkel offen ist und die Abdeckung des Trabekelwerks durch periphere anteriore Synechien nicht ausgedehnt ist, aber die Erfolgsrate ist niedriger als bei PCG. Bei Versagen der Kammerwinkelchirurgie können Trabekulektomie oder Tubus-Shunt-Operation mit Platte die erste Wahl sein. 4)
Bei der Peters-Anomalie erfolgt die Behandlung analog zum primären kongenitalen Glaukom (PCG), jedoch erreicht nur etwa ein Drittel der operierten Fälle einen guten postoperativen Augeninnendruck, und die Prognose ist häufig ungünstig. Aufgrund begleitender Hornhautanomalien ist es oft schwierig, eine praktikable Sehschärfe zu erreichen. 4)
Das mit Aniridie assoziierte Glaukom wird ebenfalls nach den Richtlinien des PCG behandelt. 4)
Peters-Anomalie: In leichten Fällen nimmt die Hornhauttrübung oft allmählich ab. Bei normalem Augeninnendruck kommt es häufig zu einer gewissen Besserung, und da die Prognose nach einer Hornhauttransplantation schlecht ist, wird eine Hornhauttransplantation im Säuglingsalter in der Regel nicht durchgeführt. Viele Fälle sind resistent gegen die medikamentöse Behandlung des Glaukoms und auch nach einer Abflusswegsrekonstruktion schwer zu kontrollieren, mit einer schlechten Prognose.
CHED: Bei Hornhautendothelinsuffizienz kann eine Hornhauttransplantation (einschließlich Endotheltransplantation) indiziert sein.
Skleralisierte Hornhaut: Kann auch mit anderen ASD-Syndromen einhergehen; schwere Fälle können eine Hornhauttransplantation erfordern.
Photophobie und Asthenopie : Verwendung von Sonnenbrillen und Verordnung von kosmetischen Kontaktlinsen in Betracht ziehen.
Aniridie-assoziierte Keratopathie (AAK) : Bei fortschreitender Hornhauttrübung wird eine Transplantation von Limbusstammzellen der Hornhaut in Betracht gezogen.
Wilms-Tumor-Screening: Bei sporadischer Aniridie wird eine Überweisung an den Kinderarzt und ein Screening bis zum Alter von 6 Jahren empfohlen. 4)
Auch wenn der Augeninnendruck sinkt, ist oft eine Amblyopiebehandlung erforderlich. Refraktive Anisometropie, irregulärer Astigmatismus, Hornhauttrübungen und Haab-Linien können eine Amblyopie verursachen, daher sollten Seh- und Refraktionsuntersuchungen parallel zur Augeninnendruckmessung fortgesetzt werden. Das Fortschreiten der Myopie und die Verlängerung der Achsenlänge deuten auf ein Fortschreiten des Glaukoms hin, sodass regelmäßige Messungen erforderlich sind.
6. Pathophysiologie und detaillierter Entstehungsmechanismus
Die normale Bildung des vorderen Augenabschnitts folgt einem komplexen Entwicklungsprogramm. Zu Beginn der 3. Embryonalwoche bildet sich der Sehgruben (Sulcus opticus) in der Neuralplatte, was den Beginn der Entwicklung des Sehorgans darstellt. Am Ende der 3. Woche bildet sich das Augenbläschen, und in der 4. Woche der Augenbecher. Ab etwa der 6. Woche beginnt der Verschluss der embryonalen Spalte, der in der 7. Woche abgeschlossen ist. Das Mesenchym, das die Vorderseite der Linse bedeckt, trennt sich und bildet die Vorderkammer.
Neuralleistenzellen de-epithelialisieren von der Neuralleiste und wandern nach einer epithelial-mesenchymalen Transition in verschiedene Bereiche des Auges. Trabekelzellen stammen von der Neuralleiste ab, während das juxtakanalikuläre Bindegewebe von vaskulären Endothelzellen abstammt; dieser Ursprungsunterschied bildet den Ort des größten Widerstands für den Kammerwasserabfluss.
PAX6 : „Master-Regulator“ der Augenentwicklung. Auf Chromosom 11 lokalisiert. Beteiligt an Aniridie, Peters-Anomalie und kongenitalem Iris-Ektropion-Syndrom.
PITX2 : Transkriptionsfaktor. Chromosom 4 (4q25). Mit sowohl okulären als auch auditiven Symptomen bei ARS assoziiert.
FOXC1 : Transkriptionsfaktor. Chromosom 6 (6p25). An ARS beteiligt, assoziiert mit sowohl okulären als auch auditiven Symptomen wie PITX2.
CYP1B1 : Cytochrom-P450-Familienenzym (GLC3A-Locus). Beteiligt an primärem kongenitalem Glaukom, Peters-Anomalie und Sklerokornea.
CHRDL1 : Beteiligt an der Entwicklung von Hornhautstroma und -endothel. Gen für X-chromosomale Megalokornea.
B3GLCT : Gen für Peters-Plus-Syndrom. Mit Glykosylierungsdefekt assoziiert. Autosomal-rezessiver Erbgang.
Das sekundäre Glaukom bei ASDA entsteht hauptsächlich durch eine Dysgenesie des Kammerwasserabflusstrakts. Mehrere Faktoren sind beteiligt:
Unreife des Trabekelwerks: Das juxtakanalikuläre Bindegewebe ist abnormal dick mit übermäßiger Ansammlung extrazellulärer Matrix.
Anheftung des Ziliarkörpers an das Trabekelwerk: Die Kontraktion des Ziliarmuskels zieht den Skleralsporn nach vorne und komprimiert den Schlemm-Kanal und das Trabekelwerk.
Hoher Ansatz der Iriswurzel: Die Iriswurzel liegt auf Höhe des Trabekelwerks und behindert den Abfluss des Kammerwassers.
Hypoplasie oder Fehlen des Schlemm-Kanals.
Das ICE-Syndrom hat eine andere Ätiologie als andere ASDA. Eine virale Ätiologie, bei der das Herpes-simplex-Virus (HSV) an der Degeneration der Hornhautendothelzellen beteiligt ist, wurde vorgeschlagen, ist aber nicht bestätigt. Es ist erworben und tritt bei Erwachsenen mittleren Alters (etwas häufiger bei Frauen) auf und ist in der Regel einseitig, was es von anderen ASDA unterscheidet.
Pathophysiologie der Aniridie-assoziierten Keratopathie (AAK)
Patienten mit Aniridie entwickeln im Laufe ihres Lebens eine fortschreitende Hornhauttrübung. Als Hauptmechanismus wird eine Limbusstammzellinsuffizienz (LSCD) angesehen. Mehrere Studien mit bestätigter PAX6-Mutation haben diese fortschreitende Veränderung dokumentiert. Die Inzidenz wird mit 20 bis über 80 % angegeben und tritt häufig symmetrisch auf, aber nicht immer. 2)
Durch Exom- und Genomanalysen werden neue assoziierte Gene identifiziert. In 40–75 % der Fälle ist das verursachende Gen noch nicht identifiziert, und die Analyse der verbleibenden „ungeklärten Fälle“ bleibt eine wichtige zukünftige Aufgabe. Die Aufklärung der Korrelation zwischen Genotyp und klinischem Phänotyp wird für die personalisierte Medizin erwartet.
Bei Fällen mit FOXC1- und PITX2-Mutationen variieren der Zeitpunkt des Glaukomausbruchs und das klinische Bild. Während der Genotyp mit der phänotypischen Vielfalt zusammenhängen kann, kann dieselbe Genmutation unterschiedliche Krankheitsformen hervorrufen, was Diagnose und Prognose erschwert. 1)
Die Inzidenz der Aniridie-assoziierten Keratopathie (AAK) wird mit 20–80 % oder mehr angegeben, und mehrere Studien mit bestätigter PAX6-Mutation dokumentierten eine lebenslange Progression der Hornhauttrübung. Die Forschung zur Transplantation von Limbusstammzellen, die auf LCSD abzielt, schreitet voran, befindet sich jedoch derzeit noch im Forschungsstadium und hat sich noch nicht als Standardbehandlung etabliert. 2)
Epidemiologische Studien haben einen Zusammenhang zwischen der Exposition gegenüber Luftverschmutzung (PM2.5) vor der Empfängnis und während der Schwangerschaft und dem ASDA-Risiko gezeigt, und aus umweltpräventivmedizinischer Sicht wird eine Anwendung im öffentlichen Gesundheitswesen untersucht. Dies könnte zu zukünftigen Präventionsstrategien führen.
Die Anwendung von Mikropulslasern und minimalinvasiven Glaukomchirurgie (MIGS)-Geräten bei Kindern mit ASDA befindet sich in der Forschungsphase. Langzeitdaten sind begrenzt, und eine vergleichbare Wirksamkeit und Sicherheit wie beim adulten Glaukom ist nicht etabliert.
Knight LSW, Ruddle JB, Taranath DA, et al. Childhood and Early Onset Glaucoma Classification and Genetic Profile in a Large Australasian Disease Registry. Ophthalmology. 2021;128(11):1549-1560. doi:10.1016/j.ophtha.2021.04.016.
Hu JCW, Trief D. A narrative review of limbal stem cell deficiency & severe ocular surface disease. Ann Eye Sci. 2023;8:13. doi:10.21037/aes-22-35. https://aes.amegroups.org/article/view/7385/html
Pazos M, Traverso CE, Viswanathan A; European Glaucoma Society. European Glaucoma Society - Terminology and guidelines for glaucoma, 6th Edition. Br J Ophthalmol. 2025;109(Suppl 1):1-212. doi:10.1136/bjophthalmol-2025-egsguidelines. PMID:41026937.