ARS Typ 1
Ursächliches Gen: PITX2 (4q25)
Hauptanomalien: Anomalien des vorderen Augenabschnitts, Zahnanomalien, überschüssige Haut um den Nabel / Nabelhernie, kraniofaziale Anomalien, kardiovaskuläre Anomalien

Das Axenfeld-Rieger-Syndrom (ARS) ist eine Gruppe angeborener Erkrankungen, die Fehlbildungen des vorderen Augenabschnitts mit systemischen Anomalien kombinieren. Die zugrundeliegende Ursache ist eine gestörte Migration und Differenzierung von Neuralleistenzellen. Im späten fetalen Stadium ist der normale Rückgang der undifferenzierten Endothelzellen, die die Vorderkammer auskleiden, aus der Iris und dem Kammerwinkel gestört; ihr Verbleib führt zu Strangbildungen und einer hohen Irisinsertion.
Historischer Hintergrund: 1920 beschrieb Axenfeld den hinteren embryonalen Ring (Vorverlagerung und Verdickung der Schwalbe-Linie) und Irisfortsätze. 1934–1935 ergänzte Rieger die Irishypoplasie, Pupillenverlagerung und Polykorie. Heute wird in drei Stufen eingeteilt:
Diese werden zusammenfassend als Axenfeld-Rieger-Syndrom bezeichnet. In 50–60 % der Fälle tritt ein Glaukom auf, die Vererbung ist autosomal-dominant und in der Regel beidseitig. Katarakt und Linsenektopie sind ebenfalls häufig assoziiert.
Epidemiologie: Die Prävalenz wurde früher mit etwa 1/200.000 angegeben, neuere Berichte schätzen sie jedoch auf 1/50.000 bis 1/100.0002)4). Es gibt keine Geschlechtsunterschiede, und die Diagnose wird häufig im Säuglings- oder Kleinkindalter gestellt.
Genetische Klassifikation:
ARS Typ 1
Ursächliches Gen: PITX2 (4q25)
Hauptanomalien: Anomalien des vorderen Augenabschnitts, Zahnanomalien, überschüssige Haut um den Nabel / Nabelhernie, kraniofaziale Anomalien, kardiovaskuläre Anomalien
ARS Typ 2
Ursächliches Gen: 13q14 (nicht bestätigt)
Hauptanomalien: Anomalien des vorderen Augenabschnitts, Glaukom. Systemische Anomalien sind seltener als bei Typ 1 und 3.
ARS Typ 3
Ursächliches Gen: FOXC1 (6p25)
Hauptanomalien: Anomalien des vorderen Augenabschnitts, Glaukom, sensorineuraler Hörverlust, Vorhofseptumdefekt, Nierenanomalien, Läsionen der weißen Substanz
Mutationen in FOXC1 und PITX2 machen 40–70 % der ARS-Fälle aus5). Allerdings ist bei 60 % der ARS-Fälle das ursächliche Gen nicht identifiziert4), was auf eine große genetische Vielfalt hinweist.
Eine große Registeranalyse des kindlichen und juvenilen Glaukoms ergab eine molekulare Diagnoserate von 56,5 %11). FOXC1-Mutationen machten 20,3 %, PITX2-Mutationen 17,4 % und PAX6-Mutationen 10,1 % aus, und es gibt nicht wenige Fälle, die durch bekannte Gene nicht erklärt werden können11).
Sie werden durch das ursächliche Gen unterschieden. Typ 1 wird durch eine PITX2-Mutation (4q25) verursacht und geht mit Zahn-, Nabel- und Gesichtsknochenanomalien einher. Typ 3 wird durch eine FOXC1-Mutation (6p25) verursacht und geht mit Hörverlust, Herzfehlern, Nierenanomalien und neurologischen Anomalien einher. Typ 2 liegt auf 13q14, aber das ursächliche Gen ist nicht bestätigt; er ist hauptsächlich durch Anomalien des vorderen Augenabschnitts und Glaukom gekennzeichnet. Die Diagnose kann durch Gentests bestätigt werden.
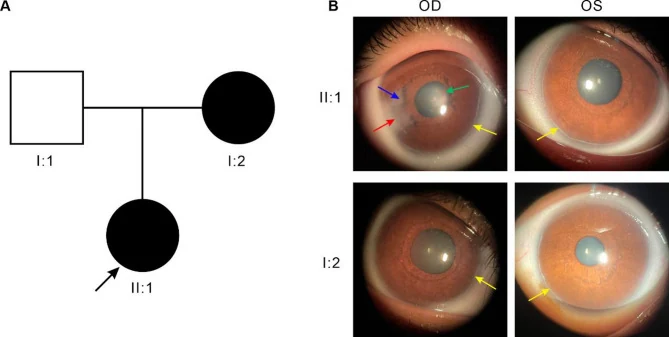
Die wichtigsten Augenbefunde sind unten aufgeführt.
| Augenbefund | Merkmale |
|---|---|
| Hinterer Embryonalring | Vorverlagerung und Verdickung der Schwalbe-Linie |
| Irisfortsätze | Fadenförmig bis breitbandig |
| Pupillenverlagerung | Verlagerung in die entgegengesetzte Richtung des hinteren Embryonalrings |
| Pseudopolykorie | Perforationsartiges Aussehen des Irisstromas |
| Uveale Eversion | Umstülpung des Irispigmentepithels |
Der hintere Embryonalring ist ein Überbleibsel undifferenzierter Zellen an der Schwalbe-Linie, das sich als lineare Struktur entlang des Limbus, 0,5–2,0 mm zentralwärts versetzt, zeigt. Er ist oft nicht zirkulär, sondern auf einen Bereich begrenzt. Wenn die prominente Schwalbe-Linie mit der Iris verklebt, spricht man von einer Axenfeld-Anomalie; wenn zusätzlich eine Atrophie des Irisstromas vorliegt, von einer Rieger-Anomalie.
Die Hornhaut ist normalerweise klar und das Endothel normal, aber durch physikalischen Kontakt mit dem Restgewebe kann sekundär eine Hornhauttrübung entstehen. Die Trübung ist oft auf die Peripherie beschränkt und beeinträchtigt in der Regel nicht direkt das Sehvermögen. Bei FOXC1-Mutationen sind Hornhauttrübung und Hornhautneovaskularisation jedoch ausgeprägter, und der Schweregrad der Hornhautanomalie ist größer als bei PITX2-Mutationen, ebenso die Häufigkeit von Glaukom1).
Die Kammerwinkelbefunde umfassen eine hohe Irisinsertion, strangförmige Uveareste und eine Verdickung der Schwalbe-Linie (hinterer Embryonalring). Es wurden auch Fälle mit Mikrosphärophakie und Linsensubluxation berichtet7).
Ein Glaukom tritt in 50–60 % der Fälle auf. Ein erhöhter Augeninnendruck kann bereits im Säuglingsalter auftreten, meistens manifestiert er sich jedoch im Kindes- oder jungen Erwachsenenalter. Manche Fälle werden aufgrund einer progredienten Sehverschlechterung diagnostiziert; es ist wichtig, die Vorderabschnittsbefunde und glaukomatösen Veränderungen nicht zu übersehen6).
Li et al. (2021) berichteten über einen 7-jährigen Jungen (ARS Typ 3, de novo FOXC1-Mutation) mit einem Hornhautdurchmesser von 14 mm, einer Achsenlänge von 27,16/26,56 mm, einem C/D-Verhältnis von 0,9 und einem IOP von 33/20 mmHg. Ab dem 36. Lebenstag war eine beidseitige antiglaukomatöse Operation erforderlich5).
Die systemischen Befunde sind wie folgt:
Etwa 50–60 % der ARS-Patienten entwickeln ein Glaukom. Es tritt häufig im Kindes- oder jungen Erwachsenenalter auf, kann aber auch bereits im Säuglingsalter mit erhöhtem Augeninnendruck einhergehen. Regelmäßige Augeninnendruckmessungen und Beurteilungen des Sehnervs sind erforderlich. Weitere Einzelheiten finden Sie im Abschnitt „Standardbehandlung“.
ARS wird autosomal-dominant vererbt mit vollständiger Penetranz. Allerdings können Personen innerhalb derselben Familie mit derselben Genmutation sehr unterschiedliche klinische Bilder aufweisen (variable Expressivität)1).
In einer großen Kohortenstudie waren FOXC1- und PITX2-Mutationen mit einem breiten Glaukomspektrum von der Kindheit bis zum Erwachsenenalter assoziiert9). Klinisch als primäres kongenitales Glaukom (PCG) diagnostizierte Fälle können durch Gentests neu klassifiziert werden. Wenn die Vorderabschnittsbefunde bei Säuglingen subtil sind, können Gentests zu einer genauen Diagnose des Krankheitstyps beitragen.
Bei Fällen mit einer Mikrodeletion in der Umgebung des PITX2-Gens kann die Überlappung der Deletionen von NEUROG2, UGT8 und NDST4 zu Entwicklungsverzögerung und geistiger Behinderung führen8)3).
Kawanami et al. (2023) berichteten über einen 3-jährigen japanischen Jungen mit einer 2,5 Mb großen Mikrodeletion in 4q25 (einschließlich PITX2, NEUROG2 und ANK2). Er zeigte eine Nabelhernie, ein Iriskolobom und eine Entwicklungsverzögerung, aber das EKG war trotz der ANK2-Deletion normal. Die Haploinsuffizienz von NEUROG2 wurde als mögliche Ursache der Entwicklungsverzögerung angesehen8).
Aufgrund des autosomal-dominanten Erbgangs beträgt die Wahrscheinlichkeit einer Vererbung von einem Elternteil mit Mutation 50 %. Die Penetranz ist vollständig, aber der Phänotyp variiert: Die Schwere der Symptome kann bei derselben Mutation stark variieren1). Gentest und genetische Beratung werden empfohlen.
Die Diagnose von ARS basiert auf beidseitigen Kammerwinkel- und Irisanomalien. Die diagnostische Bedingung ist ein hinterer Embryotoxon mit teilweiser Anheftung der peripheren Iris13). Wenn der hintere Embryotoxon nicht mit der Spaltlampe bestätigt werden kann, ist eine Gonioskopie erforderlich. Bei systemischen Anomalien wird eine vollständige pädiatrische Untersuchung im Rahmen eines ARS-Syndroms angefordert13).
Beachten Sie, dass ein leichter hinterer Embryotoxon bei 8–15 % der Normalbevölkerung vorkommt, aber allein nicht mit Glaukom usw. assoziiert ist. Die Erhebung der Familienanamnese ist ebenfalls wichtig für die Diagnose.
In der Klassifikation des pädiatrischen Glaukoms gemäß den Glaukom-Leitlinien (5. Auflage) wird ARS als typisches Beispiel für Glaukom im Zusammenhang mit angeborenen Augenentwicklungsanomalien aufgeführt13). Die Diagnose wird gestellt, wenn seit der Geburt bestehende Augenentwicklungsanomalien die Diagnosekriterien für pädiatrisches Glaukom erfüllen.
Die wichtigsten Erkrankungen, die von ARS unterschieden werden müssen, sind unten aufgeführt.
| Erkrankung | Unterschied zu ARS |
|---|---|
| ICE-Syndrom | Einseitig, erworben, weibliche Dominanz |
| Peters-Anomalie | Zentrale Hornhauttrübung, Descemet-Membran-Defekt |
| Aniridie | Hornhautpannus, Foveahypoplasie |
| Posteriore polymorphe Hornhautdystrophie | Beidseitig, familiär, kein Geschlechtsunterschied |
Das ICE-Syndrom (progressive Irisatrophie, Chandler-Syndrom usw.) muss von ARS unterschieden werden, aber der wichtigste Unterscheidungspunkt ist, dass ICE einseitig und erworben ist, während ARS beidseitig und angeboren ist.
Eine kausale Therapie der ARS selbst existiert derzeit nicht. Die Behandlung konzentriert sich auf das Glaukommanagement und die Überwachung systemischer Komplikationen. Das Behandlungsschema orientiert sich am frühkindlichen Glaukom (primäres kongenitales Glaukom: PCG) 13).
Ein Glaukom tritt bei etwa 50–60 % der ARS-Fälle auf. Die medikamentöse Behandlung erfolgt analog zum allgemeinen Glaukom, ist jedoch oft unwirksam.
Kammerwasserproduktionshemmer
Betablocker : Eines der Mittel der ersten Wahl. Sicher und wirksam, aber bei Kindern oft unwirksam.
Karboanhydrasehemmer (CAI) Augentropfen : Brinzolamid usw. Kombination mit Betablockern möglich.
Alpha-2-Agonisten (Brimonidin) : Bei Kindern unter 2 Jahren aufgrund neuropsychiatrischer Wirkungen (Apnoe, Bradykardie, Hypotonie, Muskelhypotonie, ZNS-Depression) kontraindiziert 13).
Kammerwasserabflussförderer
Prostaglandinanaloga : Latanoprost, Travoprost usw. Die Wirkung bei Kindern wird als schwächer als bei Erwachsenen angesehen 13).
Beispiel : Bei einem 7-jährigen Jungen Langzeitkontrolle mit Travoprost + Brinzolamid 5). Bei einem 77-jährigen Mann war der IOD trotz Latanoprost, Timolol und Brinzolamid mit 35 mmHg schwer kontrollierbar 2).
Einem Bericht zufolge besteht kein Unterschied in der Wirksamkeit zwischen Prostanoid-FP-Rezeptoragonisten und Betablockern 13).
Bei Säuglingen ist die Dosis der Augentropfen im Verhältnis zu Körpergewicht und Körperoberfläche relativ hoch, daher sollten möglichst niedrig konzentrierte Medikamente verwendet werden 13).
Wenn der Augeninnendruck medikamentös nicht kontrolliert werden kann, wird eine Operation durchgeführt 10)13).
Zu den berichteten Komplikationen nach GDD gehören: flache Vorderkammer 13,6 %, Hypotonie 11,7 %, Aderhauterguss 8,3 %, Endophthalmitis 1,7 % 14).
Chakraborty et al. (2022) berichteten über einen Fall einer Netzhautablösung im Zusammenhang mit Axenfeld-Rieger-Syndrom (15-jähriger Junge). Es bestanden Mikrosphärophakie und Linsensubluxation; nach Vitrektomie stieg der IOD auf 41 mmHg und es bildete sich ein Sklerastaphylom. Eine Dioden-Zyklophotokoagulation wurde durchgeführt, die schließlich zu einem IOD von 18 mmHg führte 7).
Die Erfolgsrate der Kammerwinkelchirurgie ist niedriger als bei PCG 13). Bei der Trabekulektomie mit MMC beträgt die langfristige Erfolgsrate nach 2 Jahren etwa 59 %; bei GDD werden 87 % nach 12 Monaten und 77 % nach 24 Monaten berichtet 14). In therapierefraktären Fällen können mehrere Operationen erforderlich sein.
Die grundlegende Ursache von ARS ist ein Defekt in der Migration und Differenzierung von Neuralleistenzellen. Eine gestörte Entwicklung der Neuralleistenzellen in der Vorderkammer, dem Kammerwinkel des vorderen Augenabschnitts, den Gesichtsknochen, Zähnen, dem Herz-Kreislauf-System und der periumbilikalen Haut führt zu Fehlbildungen mehrerer Organe.
Im späten fetalen Stadium verschwinden die undifferenzierten Endothelzellen, die die Vorderkammer bedecken, normalerweise aus der Iris und dem Kammerwinkel. Bei ARS ist dieser Verschwindensprozess gestört, und undifferenzierte Endothelzellen verbleiben auf der Iris, was zur Bildung von Strängen führt. Im Kammerwinkel kommt es zu einem hohen Irisansatz, der das Trabekelwerk mechanisch bedeckt.
Histologisch zeigt sich eine abnorme Ausbreitung einer einschichtigen Endothelzellschicht mit einer descemetähnlichen Membran von der hinteren Hornhautfläche über die Vorderkammer, den Kammerwinkel und die Irisoberfläche. Die Membran ist in Quadranten mit Uveaektropium und Pupillenverziehung vorhanden, während in den gegenüberliegenden Quadranten eine Irisatrophie zu beobachten ist.
FOXC1 und PITX2 sind beide Transkriptionsfaktoren, die an spezifische DNA-Sequenzen binden, um die Expression nachgeschalteter Gene zu regulieren. Beide wirken synergistisch bei der Entwicklung des vorderen Augenabschnitts und regulieren gemeinsame nachgeschaltete Zielgene 3). Die Forkhead-Domäne von FOXC1 (eine DNA-bindende Domäne aus 110 Aminosäuren) ist funktionell am wichtigsten 2), und Mutationen in dieser Domäne werden vermutlich stärker mit neuropsychiatrischen Symptomen in Verbindung gebracht.
Zwei Mechanismen der Augeninnendruckerhöhung werden genannt.
Das Ausmaß des Irisdefekts und die Menge der Irisfortsätze im Kammerwinkel korrelieren nicht unbedingt mit dem Schweregrad des Glaukoms. Ein hohes Maß an Irisverwachsungen im Kammerwinkel begünstigt jedoch die Entstehung eines Glaukoms.
FOXC1-Mutationen fördern das angeborene Glaukom stärker als andere Mutationen 1), und morphologische Anomalien des Ziliarkörpers und des Abflusswinkels können zur Erhöhung des Augeninnendrucks beitragen 1).
Bei FOXC1-Mutantenmäusen werden eine Abnahme und strukturelle Anomalien der Kollagenfasern im Hornhautstroma sowie eine Schädigung der Hornhautstromazellen beobachtet 1). Darüber hinaus fungiert FOXC1 als Inhibitor der Hornhautneovaskularisation (durch Kontrolle der Bioverfügbarkeit von VEGF) 1); der Verlust dieser Hemmung durch die FOXC1-Mutation führt zu Hornhautneovaskularisation.
FOXC1 spielt als Transkriptionsfaktor der FOX-Familie auch eine wichtige Rolle in der Gehirnentwicklung 4).
Ein systematischer Review berichtete, dass bei 41,3 % der ARS-Fälle Anomalien der weißen Substanz auftreten 4). FOXC1-Mutationen können eine zerebrale Mikroangiopathie (CSVD), Hyperintensitäten der weißen Substanz, erweiterte perivaskuläre Räume, Mikroblutungen und lakunäre Infarkte auslösen.
Ohkubo et al. (2025) bestätigten mittels MRT bei einem 2-jährigen japanischen Jungen (FOXC1-Mutation: c.240del, p.Y81Ifs21) periventrikuläre Läsionen der weißen Substanz, erweiterte perivaskuläre Räume und eine Tortuositas-Dilatation der Arteria vertebrobasilaris. Der Vater hatte im Alter von 18 Jahren einen Hirninfarkt erlitten 4).
Ein Review von 95 FOXC1-Mutationsfällen ergab bei 6,3 % neuropsychiatrische Symptome (Lernschwierigkeiten, Epilepsie, geistige Behinderung, Eifersuchtswahn usw.), und 83,3 % der Fälle mit Forkhead-Domänen-Mutation zeigten neuropsychiatrische Symptome 2).
Ja. Ein systematischer Review zeigt, dass bei etwa 41 % der ARS-Fälle mit FOXC1-Mutation Anomalien der weißen Substanz auftreten 4). FOXC1-Mutationen könnten mit einem Risiko für zerebrale Mikroangiopathie und Schlaganfall verbunden sein; daher ist eine langfristige neurologische Nachsorge wichtig, insbesondere bei FOXC1-Mutations-ARS.
In den letzten Jahren wurden durch Next-Generation-Sequencing und Whole-Genome-Sequencing viele neue Mutationen berichtet.
Wowra et al. (2024) identifizierten bei drei polnischen Schwestern mit ARS eine große Deletion (neue Mutation), die einen Teil von FOXC1-Exon 1 und die gesamte 3’UTR umfasst. Die Phänotypen innerhalb derselben Familie waren sehr unterschiedlich, und zunächst wurde fälschlicherweise das Chandler-Syndrom diagnostiziert 1).
Jiang et al. (2024) identifizierten in einer chinesischen Familie mit ARS Typ 1 eine komplexe genomische Umlagerung bestehend aus einer 6,15 Mb großen Deletion auf Chromosom 4q25, die PITX2 enthält, einer 45,71 Mb großen Inversion und einer 14 bp großen Deletion. Ein 11-jähriges Mädchen hatte einen IOD von 43,5/44,0 mmHg 3).
Weitere Berichte über neue Mutationen umfassen FOXC1 p.Phe136Leu (Forkhead-Domäne) 2), FOXC1 p.S82R (De-novo-Mutation) 5) und FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino et al. (2024) berichteten über einen 77-jährigen japanischen Mann mit ARS Typ 3. Ab dem 72. Lebensjahr traten Eifersuchtswahn auf, und eine Leukenzephalopathie wurde bestätigt. Eine Literaturübersicht über 95 Fälle mit FOXC1-Mutation ergab, dass 6,3 % (6/95) neuropsychiatrische Symptome aufwiesen, von denen 83,3 % (5/6) eine Forkhead-Domänen-Mutation hatten 2).
Diese Erkenntnis deutet darauf hin, dass die funktionelle Domäne der FOXC1-Mutation an der Manifestation neuropsychiatrischer Symptome beteiligt sein könnte, was die Bedeutung einer langfristigen Nachsorge aus psychischer Gesundheitssicht unterstreicht.
Es wurde darauf hingewiesen, dass FOXC1-Mutationen eine CSVD induzieren und das Schlaganfallrisiko erhöhen können 4). Die Prävention und frühzeitige Intervention neurovaskulärer Erkrankungen bei ARS-Patienten gelten als zukünftige Forschungsthemen.
Das Verständnis der Pathologie der Hornhaut-„Skleralisierung“ durch FOXC1-Mutationen schreitet voran 1). Es wird erwartet, dass das Verständnis der molekularen Mechanismen der Hornhauttrübung zur Entwicklung neuer Therapien mit Gentherapie, antifibrotischen Medikamenten und Biomaterialien führen kann 1).