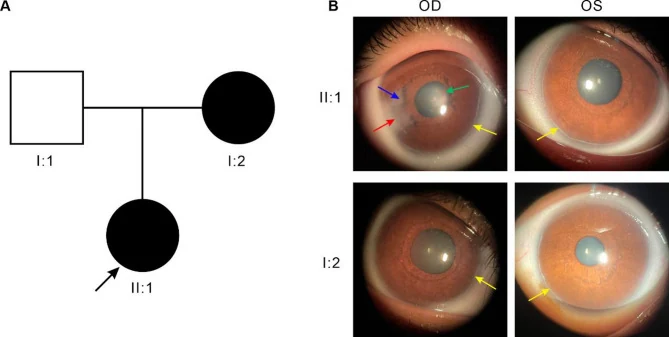
ARS ชนิดที่ 1
ยีนที่เป็นสาเหตุ: PITX2 (4q25)
ความผิดปกติหลัก: ความผิดปกติของส่วนหน้าของตา, ความผิดปกติของฟัน, ผิวหนังส่วนเกินรอบสะดือ/ไส้เลื่อนสะดือ, ความผิดปกติของกะโหลกศีรษะและใบหน้า, ความผิดปกติของหัวใจและหลอดเลือด

กลุ่มอาการแอ็กเซนเฟลด์-รีเกอร์ (ARS) เป็นกลุ่มโรคแต่กำเนิดที่รวมความผิดปกติของส่วนหน้าตาและความผิดปกติทั่วร่างกาย สาเหตุพื้นฐานคือความผิดปกติในการเคลื่อนที่และการแบ่งตัวของเซลล์นิวรัลคริสต์ ในช่วงปลายของการตั้งครรภ์ กระบวนการหายไปของเซลล์เอนโดทีเลียมที่ยังไม่แบ่งตัวซึ่งปกคลุมช่องหน้าม่านตาจากม่านตาและมุมตาถูกรบกวน และส่วนที่เหลือทำให้เกิดการสร้างเส้นใยหรือการยึดเกาะของม่านตาที่สูง
ภูมิหลังทางประวัติศาสตร์: ในปี ค.ศ. 1920 แอ็กเซนเฟลด์บรรยายถึงวงแหวนชวาลเบอด้านหลัง (การเลื่อนไปข้างหน้าและหนาตัวของเส้นชวาลเบอ) และกระบวนการของม่านตา ในปี ค.ศ. 1934-1935 รีเกอร์รายงานเพิ่มเติมเกี่ยวกับม่านตาพร่อง การเบี่ยงเบนของรูม่านตา และหลายรูม่านตา ปัจจุบันจำแนกเป็นสามระยะ:
ทั้งหมดนี้เรียกรวมกันว่ากลุ่มอาการแอ็กเซนเฟลด์-รีเกอร์ 50-60% มีต้อหินร่วมด้วย เป็นการถ่ายทอดทางพันธุกรรมแบบออโตโซมอลโดมิแนนต์และมักเป็นทั้งสองข้าง มักเกิดร่วมกับต้อกระจกและเลนส์เคลื่อน
ระบาดวิทยา: ความชุกเดิมประมาณ 1/200,000 แต่รายงานล่าสุดประมาณ 1/50,000 ถึง 1/100,0002)4) ไม่มีความแตกต่างทางเพศ และมักได้รับการวินิจฉัยในวัยทารกหรือเด็กเล็ก
การจำแนกประเภททางพันธุกรรม มีดังนี้:
ARS ชนิดที่ 1
ยีนที่เป็นสาเหตุ: PITX2 (4q25)
ความผิดปกติหลัก: ความผิดปกติของส่วนหน้าของตา, ความผิดปกติของฟัน, ผิวหนังส่วนเกินรอบสะดือ/ไส้เลื่อนสะดือ, ความผิดปกติของกะโหลกศีรษะและใบหน้า, ความผิดปกติของหัวใจและหลอดเลือด
ARS ชนิดที่ 2
ยีนที่เป็นสาเหตุ: 13q14 (ยังไม่ยืนยัน)
ความผิดปกติหลัก: ความผิดปกติของส่วนหน้าของตา, ต้อหิน ความผิดปกติทั่วร่างกายน้อยกว่าชนิดที่ 1 และ 3
ARS ชนิดที่ 3
ยีนที่เป็นสาเหตุ: FOXC1 (6p25)
ความผิดปกติหลัก: ความผิดปกติของส่วนหน้าของตา, ต้อหิน, การสูญเสียการได้ยินแบบประสาทรับเสียง, ผนังกั้นหัวใจห้องบนบกพร่อง, ความผิดปกติของไต, รอยโรคของสารสีขาว
การกลายพันธุ์ของ FOXC1 และ PITX2 คิดเป็น 40–70% ของผู้ป่วย ARS5) อย่างไรก็ตาม ใน 60% ของผู้ป่วย ARS ยังไม่ทราบยีนที่เป็นสาเหตุ4) ซึ่งแสดงถึงความหลากหลายทางพันธุกรรมที่มาก
ในการวิเคราะห์ทะเบียนผู้ป่วยต้อหินที่เริ่มในวัยเด็กและวัยหนุ่มสาวขนาดใหญ่ อัตราการวินิจฉัยระดับโมเลกุลอยู่ที่ 56.5%11) การกลายพันธุ์ของ FOXC1 คิดเป็น 20.3%, PITX2 17.4% และ PAX6 10.1% และยังมีหลายกรณีที่ไม่สามารถอธิบายได้ด้วยยีนที่รู้จัก11)
พวกมันแตกต่างกันที่ยีนที่เป็นสาเหตุ ชนิดที่ 1 เกิดจากการกลายพันธุ์ของ PITX2 (4q25) และมีความผิดปกติของฟัน สะดือ และกระดูกใบหน้าร่วมด้วย ชนิดที่ 3 เกิดจากการกลายพันธุ์ของ FOXC1 (6p25) และมีความผิดปกติของการได้ยิน หัวใจบกพร่อง ไตผิดปกติ และความผิดปกติทางระบบประสาทร่วมด้วย ชนิดที่ 2 อยู่บนโครโมโซม 13q14 แต่ยังไม่ทราบยีนที่แน่ชัด โดยมีความผิดปกติของส่วนหน้าของตาและต้อหินเป็นลักษณะสำคัญ การวินิจฉัยสามารถยืนยันได้ด้วยการตรวจทางพันธุกรรม
รายการหลักของผลการตรวจตา แสดงไว้ด้านล่าง
| ผลการตรวจตา | ลักษณะเฉพาะ |
|---|---|
| วงแหวนตัวอ่อนส่วนหลัง | เส้น Schwalbe เลื่อนไปข้างหน้าและหนาตัวขึ้น |
| ส่วนยื่นของม่านตา | เส้นใยละเอียดถึงแถบกว้าง |
| การเบี่ยงเบนของรูม่านตา | เบี่ยงเบนไปในทิศทางตรงกันข้ามกับวงแหวนตัวอ่อนส่วนหลัง |
| ภาวะรูม่านตาเทียมหลายรู | ลักษณะคล้ายรูพรุนในสโตรมาของม่านตา |
| การพลิกกลับของยูเวีย | การพลิกกลับของเยื่อบุผิวเม็ดสีม่านตา |
วงแหวนตัวอ่อนส่วนหลัง คือเซลล์ที่ยังไม่แยกตัวเหลืออยู่ที่เส้นชวาลเบอ (Schwalbe line) ปรากฏเป็นเส้นตามแนวลิมบัส ห่างจากลิมบัสเข้าด้านใน 0.5–2.0 มม. มักเป็นบางส่วนไม่เต็มรอบ หากเส้นชวาลเบอที่ยื่นออกมายึดติดกับม่านตา เรียกว่า Axenfeld anomaly หากมีฝ่อของสโตรมาม่านตาร่วมด้วย เรียกว่า Rieger anomaly
กระจกตา ปกติใสและโครงสร้างเอนโดทีเลียมปกติ แต่การสัมผัสทางกายภาพกับเนื้อเยื่อที่เหลืออยู่อาจทำให้กระจกตาขุ่น secondary ได้ การขุ่นมักจำกัดอยู่บริเวณรอบนอกและโดยทั่วไปไม่ส่งผลต่อการมองเห็นโดยตรง อย่างไรก็ตาม ในกรณีกลายพันธุ์ FOXC1 การขุ่นและการสร้างเส้นเลือดใหม่ในกระจกตาจะเด่นชัดกว่า และระดับความผิดปกติของกระจกตาและความถี่ของต้อหินสูงกว่าเมื่อเทียบกับการกลายพันธุ์ PITX21)
ผลการตรวจมุมตา พบการเกาะของม่านตาสูง มีเศษยูเวียเป็นเส้น และเส้นชวาลเบอหนาตัว (วงแหวนตัวอ่อนส่วนหลัง) มีรายงานกรณีที่มีเลนส์ตารูป小球 (microspherophakia) หรือเลนส์ตาย่อยหลุดร่วมด้วย7)
ต้อหิน เกิดร่วมใน 50–60% ของกรณี ความดันลูกตาสูงอาจเกิดขึ้นตั้งแต่ทารก แต่ส่วนใหญ่เกิดในวัยเด็กถึงวัยหนุ่มสาว บางกรณีได้รับการวินิจฉัยหลังการมองเห็นลดลงแบบค่อยเป็นค่อยไป ดังนั้นจึงสำคัญที่จะไม่พลาดผลการตรวจส่วนหน้าของตาและการเปลี่ยนแปลงของต้อหิน6)
ในกรณีของ Li et al. (2021) เด็กชายอายุ 7 ปี (ARS type 3, การกลายพันธุ์ de novo FOXC1) มีเส้นผ่านศูนย์กลางกระจกตา 14 มม. ความยาวแกนตา 27.16/26.56 มม. อัตราส่วน C/D 0.9 ความดันลูกตา 33/20 มม.ปรอท ตาทั้งสองข้างต้องผ่าตัดต้อหินตั้งแต่วันที่ 36 หลังคลอด5)
ผลการตรวจทั่วร่างกาย มีดังนี้:
ประมาณ 50-60% ของผู้ป่วย ARS เกิดโรคต้อหิน มักเริ่มในวัยเด็กถึงวัยผู้ใหญ่ตอนต้น แต่ก็มีกรณีที่ความดันลูกตาสูงตั้งแต่ทารก จำเป็นต้องวัดความดันลูกตาเป็นประจำและประเมินเส้นประสาทตา สำหรับรายละเอียด โปรดดูส่วน “วิธีการรักษามาตรฐาน”
ARS มีรูปแบบการถ่ายทอดทางพันธุกรรมแบบ ** autosomal dominant** โดยมีความสมบูรณ์ในการแสดงออก (penetrance) สมบูรณ์ อย่างไรก็ตาม แม้ในครอบครัวเดียวกันที่มีการกลายพันธุ์ของยีนเดียวกัน ก็อาจมีความแตกต่างทางคลินิกอย่างมาก (variable expressivity)1)
ในการศึกษาแบบกลุ่มตัวอย่างขนาดใหญ่ การกลายพันธุ์ของ FOXC1 และ PITX2 สัมพันธ์กับสเปกตรัมกว้างของโรคต้อหินตั้งแต่วัยเด็กถึงวัยผู้ใหญ่9) กรณีที่ได้รับการวินิจฉัยทางคลินิกครั้งแรกว่าเป็นโรคต้อหินแต่กำเนิดปฐมภูมิ (PCG) บางครั้งถูกจัดประเภทใหม่ผ่านการตรวจทางพันธุกรรม และการตรวจทางพันธุกรรมมีส่วนช่วยในการวินิจฉัยชนิดที่แม่นยำเมื่อผลการตรวจส่วนหน้าของตาในทารกไม่ชัดเจน
ในกรณีที่มีไมโครดีลีชันรอบยีน PITX2 การซ้อนทับของดีลีชันของ NEUROG2, UGT8 และ NDST4 อาจทำให้เกิดพัฒนาการล่าช้าและความบกพร่องทางสติปัญญา 8)3)
Kawanami et al. (2023) รายงานเด็กชายชาวญี่ปุ่นอายุ 3 ปีที่มีไมโครดีลีชันขนาด 2.5 Mb ที่ตำแหน่ง 4q25 (รวมถึง PITX2, NEUROG2 และ ANK2) เขามีไส้เลื่อนสะดือ, คอลโลโบมาของม่านตา และพัฒนาการล่าช้า แต่คลื่นไฟฟ้าหัวใจปกติแม้จะมีการดีลีชันของ ANK2 การขาดแฮพลอยน์ฟิชเชียนซีของ NEUROG2 ถือเป็นสาเหตุที่เป็นไปได้ของพัฒนาการล่าช้า 8)
เนื่องจากเป็นแบบออโตโซมอลโดมิแนนต์ ความน่าจะเป็นที่จะได้รับการกลายพันธุ์จากพ่อแม่คือ 50% การแทรกซึมสมบูรณ์แต่ฟีโนไทป์แปรผัน และความรุนแรงของอาการอาจแตกต่างกันอย่างมากแม้จะมีการกลายพันธุ์เดียวกัน 1) แนะนำให้ตรวจทางพันธุกรรมและรับคำปรึกษาทางพันธุกรรม
การวินิจฉัย ARS ขึ้นอยู่กับ ความผิดปกติของมุมตาและม่านตาในตาทั้งสองข้าง การมี posterior embryotoxon ร่วมกับการยึดเกาะบางส่วนของม่านตารอบข้างเป็นเงื่อนไขในการวินิจฉัย 13) หากไม่เห็น posterior embryotoxon ด้วยกล้องจุลทรรศน์ชนิดกรีด จำเป็นต้องตรวจ gonioscopy หากมีความผิดปกติของระบบร่างกาย ผู้ป่วยจะถูกส่งต่อไปยังกุมารแพทย์เพื่อประเมินอย่างครบถ้วนในฐานะส่วนหนึ่งของกลุ่มอาการ ARS 13)
โปรดทราบว่า 8-15% ของประชากรปกติมี posterior embryotoxon เล็กน้อย แต่เพียงอย่างเดียวไม่สัมพันธ์กับโรคต้อหินหรืออื่นๆ ประวัติครอบครัวก็มีความสำคัญในการวินิจฉัย
ในระบบการจำแนกโรคต้อหินในเด็กตามแนวทางการรักษาโรคต้อหิน (ฉบับที่ 5) ARS ถูกจัดเป็นตัวอย่างทั่วไปของ โรคต้อหินที่เกี่ยวข้องกับความผิดปกติของพัฒนาการดวงตาแต่กำเนิด 13) การวินิจฉัยจะเกิดขึ้นเมื่อความผิดปกติของตาที่มีอยู่ตั้งแต่แรกเกิดเข้าเกณฑ์การวินิจฉัยโรคต้อหินในเด็ก
โรคหลักที่ควรแยกจาก ARS แสดงไว้ด้านล่าง
| โรค | ความแตกต่างจาก ARS |
|---|---|
| กลุ่มอาการ ICE | ข้างเดียว, ภายหลัง, พบในหญิงมากกว่า |
| ความผิดปกติของปีเตอร์ส | ตาพร่ามัวตรงกลางกระจกตา, ข้อบกพร่องของเยื่อเดสเซเมต์ |
| ภาวะไม่มีม่านตา | พานนัสที่กระจกตา, ฟอฟีอาพัฒนาน้อย |
| โรคจอประสาทตาเสื่อมแบบพหุสัณฐานส่วนหลัง | สองข้าง, ทางครอบครัว, ไม่แตกต่างทางเพศ |
กลุ่มอาการ ICE (เช่น ฝ่อของม่านตาแบบค่อยเป็นค่อยไป, กลุ่มอาการแชนด์เลอร์) ต้องแยกจาก ARS แต่ข้อแตกต่างหลักคือ ICE เป็นข้างเดียวและเกิดขึ้นภายหลัง ในขณะที่ ARS เป็นสองข้างและแต่กำเนิด
ปัจจุบันยังไม่มีการรักษาที่หายขาดสำหรับ ARS เอง การจัดการ โรคต้อหิน และการเฝ้าระวังภาวะแทรกซ้อนทางระบบเป็นหัวใจสำคัญของการรักษา แนวทางการรักษาเป็นไปตามโรคต้อหินที่พัฒนาตั้งแต่แรกเริ่ม (ต้อหินแต่กำเนิดปฐมภูมิ: PCG) 13)
โรคต้อหินเกิดขึ้นในประมาณ 50-60% ของผู้ป่วย ARS การรักษาด้วยยาจะเป็นไปตามแนวทางทั่วไปของโรคต้อหิน แต่มักไม่ได้ผล
ยาที่ยับยั้งการผลิตอารมณ์ขันน้ำ
ยาเบต้าบล็อกเกอร์: หนึ่งในตัวเลือกแรก ปลอดภัยและมีประสิทธิภาพแต่มักไม่ได้ผลในเด็ก
ยาหยอดตากลุ่มยับยั้งเอนไซม์คาร์บอนิกแอนไฮเดรส (CAI): เช่น บรินโซลาไมด์ สามารถใช้ร่วมกับยาเบต้าบล็อกเกอร์ได้
ยาอัลฟา-2 อะโกนิสต์ (บริโมนิดีน): ห้ามใช้ในเด็กอายุต่ำกว่า 2 ปี เนื่องจากผลข้างเคียงทางจิตประสาท (หยุดหายใจ หัวใจเต้นช้า ความดันโลหิตต่ำ กล้ามเนื้ออ่อนแรง กดประสาทส่วนกลาง) 13)
ยาที่เพิ่มการไหลออกของอารมณ์ขันน้ำ
ยาที่คล้ายพรอสตาแกลนดิน: เช่น ลาทาโนพรอสต์ ทราโวพรอสต์ ผลในเด็กอ่อนแอกว่าผู้ใหญ่ 13)
ตัวอย่าง: ในเด็กชายอายุ 7 ปี จัดการระยะยาวด้วยทราโวพรอสต์ + บรินโซลาไมด์ 5) ในชายอายุ 77 ปี ความดันลูกตา 35 มม.ปรอทแม้ใช้ลาทาโนพรอสต์/ไทโมลอล + บรินโซลาไมด์ แสดงว่าควบคุมได้ยาก 2)
มีรายงานว่าไม่มีความแตกต่างในประสิทธิภาพระหว่างยากลุ่มอะโกนิสต์ตัวรับ FP โพรสตานอยด์และยาเบต้าบล็อกเกอร์ 13)
ในทารกและเด็กเล็ก ปริมาณยาหยอดตาค่อนข้างมากเมื่อเทียบกับน้ำหนักและพื้นที่ผิวร่างกาย ดังนั้นควรใช้ยาที่มีความเข้มข้นต่ำที่สุดเท่าที่เป็นไปได้ 13)
หากไม่สามารถควบคุมความดันลูกตาด้วยยาได้ จะทำการผ่าตัด10)13).
ภาวะแทรกซ้อนหลัง GDD ได้แก่ ช่องหน้าตื้น 13.6% ความดันลูกตาต่ำ 11.7% น้ำซึมใต้คอรอยด์ 8.3% และการติดเชื้อในลูกตา 1.7%14).
Chakraborty และคณะ (2022) รายงานกรณีจอประสาทตาลอกที่เกี่ยวข้องกับ ARS (เด็กชายอายุ 15 ปี) ร่วมกับไมโครฟาเกียและเลนส์ตาย่อย หลังการผ่าตัดวุ้นตา ความดันลูกตาเพิ่มขึ้นเป็น 41 มิลลิเมตรปรอทและเกิดสตาฟิโลมา ได้ทำการจี้ซิลิอารีบอดี้ด้วยไดโอด และในที่สุดความดันลูกตาลดลงเหลือ 18 มิลลิเมตรปรอท7).
อัตราความสำเร็จของการผ่าตัดมุมต่ำกว่าต้อหินแต่กำเนิดปฐมภูมิ 13) ในการผ่าตัด trabeculectomy ร่วมกับ MMC อัตราความสำเร็จระยะยาว 2 ปีประมาณ 59% ส่วนอุปกรณ์ระบายต้อหินรายงานที่ 87% ที่ 12 เดือนและ 77% ที่ 24 เดือน 14) กรณีดื้อต่อการรักษาอาจต้องผ่าตัดหลายครั้ง
สาเหตุพื้นฐานของ ARS คือ ความบกพร่องในการย้ายถิ่นและการแบ่งตัวของเซลล์นิวรัลครีสต์ การพัฒนาที่บกพร่องของเซลล์นิวรัลครีสต์ในช่องหน้าม่านตา มุมตา กระดูกใบหน้า ฟัน ระบบหัวใจและหลอดเลือด และผิวหนังรอบสะดือ ทำให้เกิดความผิดปกติของหลายอวัยวะ
ในช่วงปลายของการตั้งครรภ์ เซลล์เอนโดทีเลียมที่ยังไม่แยกตัวซึ่งบุช่องหน้าม่านตาปกติจะหายไปจากม่านตาและมุมตา ใน ARS กระบวนการหายไปนี้บกพร่อง และเซลล์เอนโดทีเลียมที่ยังไม่แยกตัวยังคงอยู่บนม่านตา ทำให้เกิดการสร้างแถบเนื้อเยื่อ ที่มุมตาเกิดการยึดเกาะของม่านตาสูง ซึ่งปกคลุม trabecular meshwork โดยกลไก
ทางจุลกายวิภาคศาสตร์ พบเซลล์ชั้นเดียวคล้ายเอนโดทีเลียมที่มีเยื่อคล้ายเยื่อเดสเซเมทแผ่ขยายผิดปกติจากผิวด้านหลังของกระจกตาผ่านช่องหน้าม่านตา มุมตา และผิวม่านตา เยื่อนี้พบในจตุภาคที่มีการพลิกกลับของยูเวียและการเบี่ยงเบนของรูม่านตา ในขณะที่พบม่านตาฝ่อในจตุภาคตรงข้าม
FOXC1 และ PITX2 ต่างก็เป็น transcription factor ซึ่งจับกับลำดับดีเอ็นเอจำเพาะและควบคุมการแสดงออกของยีนปลายน้ำ ทั้งสองทำงานร่วมกันในการพัฒนาส่วนหน้าของตาและควบคุมยีนเป้าหมายปลายน้ำร่วมกัน 3) Forkhead domain ของ FOXC1 (โดเมนจับดีเอ็นเอขนาด 110 กรดอะมิโน) มีความสำคัญที่สุดในเชิงหน้าที่ 2) และการกลายพันธุ์ในโดเมนนี้ถูกเสนอว่ามีความสัมพันธ์ที่แน่นแฟ้นกว่ากับอาการทางจิตประสาท
มีการระบุกลไกการเพิ่มความดันลูกตาสองประการ:
ระดับของความบกพร่องของม่านตาและปริมาณของติ่งม่านตาที่มุมตาไม่จำเป็นต้องสัมพันธ์กับความรุนแรงของโรคต้อหิน อย่างไรก็ตาม การยึดติดของม่านตาที่มุมตาอย่างรุนแรงจะเพิ่มความเสี่ยงต่อโรคต้อหิน
การกลายพันธุ์ของ FOXC1 ส่งเสริมโรคต้อหินแต่กำเนิดมากกว่าการกลายพันธุ์อื่นๆ 1) และความผิดปกติทางสัณฐานวิทยาของซิลิอารีบอดีและมุมระบายอาจมีส่วนทำให้ความดันลูกตาสูงขึ้น 1)
ในหนูที่มีการกลายพันธุ์ FOXC1 พบว่ามีการลดลงของเส้นใยคอลลาเจนในสโตรมาของกระจกตา ความผิดปกติของโครงสร้าง และความเสียหายของเซลล์สโตรมา 1) นอกจากนี้ FOXC1 ยังทำหน้าที่เป็นปัจจัยยับยั้งการสร้างเส้นเลือดใหม่ในกระจกตา (ผ่านการควบคุมความพร้อมใช้ของ VEGF) 1) และการสูญเสียการยับยั้งนี้เนื่องจากการกลายพันธุ์ FOXC1 ทำให้เกิดการสร้างเส้นเลือดใหม่ในกระจกตา
FOXC1 ในฐานะปัจจัยการถอดรหัสในตระกูล FOX ยังมีบทบาทสำคัญในการพัฒนาสมอง 4)
ในการทบทวนวรรณกรรมอย่างเป็นระบบ มีรายงานว่าความผิดปกติของสารสีขาวปรากฏใน 41.3% ของผู้ป่วย ARS 4) การกลายพันธุ์ FOXC1 สามารถเหนี่ยวนำให้เกิดโรคหลอดเลือดสมองขนาดเล็ก ภาวะสัญญาณสูงของสารสีขาว การขยายของช่องรอบหลอดเลือด การตกเลือดขนาดเล็ก และภาวะสมองตายแบบลาคูนาร์
Ohkubo และคณะ (2025) ยืนยันรอยโรคของสารสีขาวรอบโพรงสมอง การขยายของช่องรอบหลอดเลือด และการคดเคี้ยวและขยายของหลอดเลือดแดงเวอร์ทีโบรบาซิลาร์ในการตรวจ MRI สมองของเด็กชายชาวญี่ปุ่นอายุ 2 ปี (การกลายพันธุ์ FOXC1: c.240del, p.Y81Ifs21) บิดาของเขามีประวัติสมองตายเมื่ออายุ 18 ปี 4)
ในการทบทวนผู้ป่วยที่มีการกลายพันธุ์ FOXC1 จำนวน 95 ราย พบอาการทางจิตประสาท (ความยากลำบากในการเรียนรู้ โรคลมชัก ภาวะบกพร่องทางสติปัญญา อาการหลงผิดหึงหวง ฯลฯ) ใน 6.3% และอาการทางจิตประสาทปรากฏใน 83.3% ของผู้ป่วยที่มีการกลายพันธุ์ในโดเมน forkhead 2)
ใช่ มีการทบทวนวรรณกรรมอย่างเป็นระบบรายงานว่าความผิดปกติของสารสีขาวปรากฏในประมาณ 41% ของผู้ป่วย ARS ที่มีการกลายพันธุ์ FOXC1 4) การกลายพันธุ์ FOXC1 อาจเกี่ยวข้องกับโรคหลอดเลือดสมองขนาดเล็กและความเสี่ยงต่อโรคหลอดเลือดสมอง โดยเฉพาะใน ARS ชนิด FOXC1 การติดตามทางระบบประสาทในระยะยาวเป็นสิ่งสำคัญ
ในช่วงไม่กี่ปีที่ผ่านมา มีรายงานการกลายพันธุ์ใหม่จำนวนมากผ่านการหาลำดับยุคถัดไปและการหาลำดับจีโนมทั้งหมด
Wowra และคณะ (2024) ระบุการขาดหายขนาดใหญ่ (การกลายพันธุ์ใหม่) ซึ่งรวมถึงส่วนหนึ่งของเอ็กซอน 1 และ 3’UTR ทั้งหมดของ FOXC1 ในพี่น้องสาวชาวโปแลนด์สามคนที่เป็น ARS ฟีโนไทป์แตกต่างกันอย่างมากแม้ในครอบครัวเดียวกัน และในตอนแรกได้รับการวินิจฉัยผิดว่าเป็นกลุ่มอาการแชนด์เลอร์ 1)
Jiang และคณะ (2024) ระบุการจัดเรียงจีโนมที่ซับซ้อนซึ่งประกอบด้วยการขาดหาย 6.15 Mb บนโครโมโซม 4q25 ที่รวม PITX2 การกลับด้าน 45.71 Mb และการขาดหาย 14 bp ในครอบครัวชาวจีนที่เป็น ARS ชนิดที่ 1 เด็กหญิงอายุ 11 ปีมีความดันลูกตา 43.5/44.0 มิลลิเมตรปรอท 3)
รายงานการกลายพันธุ์ใหม่อื่นๆ ได้แก่ FOXC1 p.Phe136Leu (โดเมน forkhead) 2), FOXC1 p.S82R (การกลายพันธุ์ใหม่) 5) และ FOXC1 c.240del, p.Y81Ifs21 4)
Yoshino และคณะ (2024) รายงานกรณีชายชาวญี่ปุ่นอายุ 77 ปีที่เป็น ARS ชนิดที่ 3 อาการหลงผิดหึงหวงปรากฏตั้งแต่อายุ 72 ปี และได้รับการยืนยันว่าเป็นโรคเม็ดเลือดขาวผิดปกติ ในการทบทวนวรรณกรรม 95 กรณีของการกลายพันธุ์ FOXC1 พบอาการทางจิตประสาทใน 6.3% (6/95) และ 83.3% (5/6) ในจำนวนนี้มีการกลายพันธุ์ในโดเมน forkhead 2)
การค้นพบนี้ชี้ให้เห็นว่าโดเมนหน้าที่ของการกลายพันธุ์ FOXC1 อาจเกี่ยวข้องกับการแสดงอาการทางจิตประสาท ซึ่งบ่งชี้ถึงความสำคัญของการติดตามผลระยะยาวจากมุมมองด้านสุขภาพจิต
มีการชี้ให้เห็นว่าการกลายพันธุ์ FOXC1 อาจเหนี่ยวนำให้เกิด CSVD และเพิ่มความเสี่ยงต่อโรคหลอดเลือดสมอง 4) การป้องกันและการแทรกแซงตั้งแต่เนิ่นๆ สำหรับโรคหลอดเลือดประสาทในผู้ป่วย ARS เป็นหัวข้อวิจัยในอนาคต
ความเข้าใจเกี่ยวกับพยาธิกำเนิดของ “การกลายเป็นเส้นโลหิต” ของกระจกตาที่เกิดจากการกลายพันธุ์ FOXC1 กำลังก้าวหน้า 1) ความเข้าใจเกี่ยวกับกลไกระดับโมเลกุลของความขุ่นของกระจกตาคาดว่าจะนำไปสู่การพัฒนาการรักษาใหม่โดยใช้ยีนบำบัด ยาต้านการเกิดพังผืด และวัสดุชีวภาพ 1)