ARS نوع 1
ژن عامل: PITX2 (4q25)
ناهنجاریهای اصلی: ناهنجاریهای بخش قدامی چشم، ناهنجاریهای دندانی، پوست اضافی دور ناف/فتق نافی، ناهنجاریهای جمجمهای-صورتی، ناهنجاریهای قلبی-عروقی

سندرم آکسنفلد-ریگر (ARS) گروهی از بیماریهای مادرزادی است که ناهنجاریهای بخش قدامی چشم و ناهنجاریهای سیستمیک را ترکیب میکند. علت اصلی آن اختلال در مهاجرت و تمایز سلولهای تاج عصبی است. در اواخر دوره جنینی، روند ناپدید شدن طبیعی سلولهای اندوتلیال تمایزنیافته پوشاننده اتاق قدامی از عنبیه و زاویه مختل میشود و باقیماندن آنها باعث ایجاد طنابهای فیبری و چسبندگی بالای عنبیه میشود.
پیشینه تاریخی: در سال 1920، آکسنفلد حلقه جنینی خلفی (جابجایی و ضخیم شدن خط اشوالبه) و زوائد عنبیه را توصیف کرد. در سالهای 1934-1935، ریگر هیپوپلازی عنبیه، انحراف مردمک و پلی کوریا را گزارش کرد. امروزه این بیماری در سه مرحله طبقهبندی میشود:
همه این موارد تحت عنوان سندرم آکسنفلد-ریگر نامیده میشوند. 50 تا 60 درصد موارد با گلوکوم همراه است و به صورت اتوزومال غالب و معمولاً دوطرفه منتقل میشود. آب مروارید و جابجایی عدسی نیز با شیوع بالا همراه هستند.
اپیدمیولوژی: شیوع حدود 1/200,000 تخمین زده شده است، اما گزارشهای اخیر 1/50,000 تا 100,000 را پیشنهاد میکنند2)4). تفاوت جنسیتی ندارد و اغلب در دوران نوزادی یا کودکی تشخیص داده میشود.
طبقهبندی ژنتیکی به شرح زیر است:
ARS نوع 1
ژن عامل: PITX2 (4q25)
ناهنجاریهای اصلی: ناهنجاریهای بخش قدامی چشم، ناهنجاریهای دندانی، پوست اضافی دور ناف/فتق نافی، ناهنجاریهای جمجمهای-صورتی، ناهنجاریهای قلبی-عروقی
ARS نوع 2
ژن عامل: 13q14 (تأیید نشده)
ناهنجاریهای اصلی: ناهنجاریهای بخش قدامی چشم، گلوکوم. ناهنجاریهای سیستمیک کمتر از نوع 1 و 3 است.
ARS نوع 3
ژن عامل: FOXC1 (6p25)
ناهنجاریهای اصلی: ناهنجاریهای بخش قدامی چشم، گلوکوم، کمشنوایی حسی-عصبی، نقص دیواره بیندهلیزی، ناهنجاری کلیوی، ضایعات ماده سفید
جهشهای FOXC1 و PITX2 40-70% موارد ARS را تشکیل میدهند5). با این حال، در 60% موارد ARS ژن عامل ناشناخته است4) و تنوع ژنتیکی قابل توجه است.
در یک تحلیل رجیستری بزرگ از گلوکوم دوران کودکی و جوانی، نرخ تشخیص مولکولی 56.5% بود11). جهش FOXC1 20.3%، PITX2 17.4% و PAX6 10.1% موارد را تشکیل میدادند و موارد غیرقابل توضیح با ژنهای شناخته شده کم نیستند11).
آنها بر اساس ژن عامل تفکیک میشوند. نوع 1 با جهش PITX2 (4q25) همراه با ناهنجاریهای دندانی، نافی و استخوانهای صورت است. نوع 3 با جهش FOXC1 (6p25) همراه با کمشنوایی، نقص قلبی، ناهنجاری کلیوی و عصبی است. نوع 2 در 13q14 قرار دارد اما ژن عامل تأیید نشده و عمدتاً با ناهنجاری بخش قدامی چشم و گلوکوم مشخص میشود. تشخیص قطعی با آزمایش ژنتیک امکانپذیر است.
موارد اصلی یافتههای چشمی در زیر آورده شده است.
| یافته چشمی | ویژگی |
|---|---|
| حلقه جنینی خلفی | جابهجایی و ضخیمشدگی خط اشوالبه به سمت جلو |
| برآمدگیهای عنبیه | نخی شکل تا نواری پهن |
| انحراف مردمک | انحراف به سمت مخالف حلقه جنینی خلفی |
| چندمردمکی کاذب | ظاهر سوراخ مانند در استرومای عنبیه |
| اورژن یووهآ | برگشتگی اپیتلیوم رنگدانهدار عنبیه |
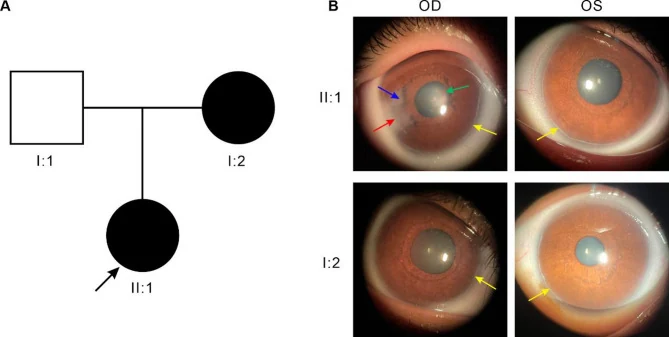
حلقه جنینی خلفی باقیماندن سلولهای تمایزنیافته در ناحیه خط اشوالبه است که به صورت خطی در امتداد لیمبوس، 0.5 تا 2.0 میلیمتر به سمت مرکز از لیمبوس دیده میشود. اغلب به جای تمام محیط، در بخشی محدود میشود. اگر خط اشوالبه برجسته با عنبیه چسبندگی داشته باشد، ناهنجاری آکسنفلد و اگر با آتروفی استرومای عنبیه همراه باشد، ناهنجاری ریگر نامیده میشود.
قرنیه معمولاً شفاف و ساختار اندوتلیال طبیعی است، اما تماس فیزیکی با بافت باقیمانده میتواند ثانویه باعث کدورت قرنیه شود. کدورت قرنیه اغلب به بخش محیطی محدود میشود و معمولاً مستقیماً بر بینایی تأثیر نمیگذارد. با این حال، در جهش FOXC1، کدورت قرنیه و عروقی شدن قرنیه بارزتر است و در مقایسه با جهش PITX2، شدت ناهنجاری قرنیه بیشتر و فراوانی گلوکوم بالاتر است 1).
یافتههای زاویه شامل اتصال بالای عنبیه، بقایای طنابی یووهآ، و ضخیم شدن خط اشوالبه (حلقه جنینی خلفی) است. مواردی از عدسی کروی کوچک و سابلوکساسیون عدسی نیز گزارش شده است 7).
گلوکوم در 50 تا 60٪ موارد همراه است. افزایش فشار داخل چشم ممکن است از دوران نوزادی رخ دهد، اما بیشتر در دوران کودکی تا جوانی بروز میکند. مواردی وجود دارد که با کاهش پیشرونده بینایی تشخیص داده میشود، بنابراین مهم است که یافتههای بخش قدامی و تغییرات گلوکوماتوز نادیده گرفته نشوند 6).
در مورد پسر 7 ساله Li و همکاران (2021) (ARS نوع 3، جهش de novo FOXC1)، قطر قرنیه 14 میلیمتر، طول محوری 27.16/26.56 میلیمتر، نسبت C/D 0.9، IOP 33/20 میلیمتر جیوه بود. از روز 36 پس از تولد، جراحی ضد گلوکوم در هر دو چشم لازم بود 5).
یافتههای سیستمیک به شرح زیر است:
حدود ۵۰ تا ۶۰ درصد از افراد مبتلا به ARS به گلوکوم مبتلا میشوند. گلوکوم اغلب در دوران کودکی تا جوانی بروز میکند، اما در برخی موارد از دوران نوزادی افزایش فشار داخل چشم دیده میشود. اندازهگیری منظم فشار چشم و ارزیابی عصب بینایی ضروری است. برای جزئیات بیشتر به بخش «روشهای درمانی استاندارد» مراجعه کنید.
ARS به صورت اتوزومال غالب به ارث میرسد و نفوذ کامل دارد. با این حال، حتی در یک خانواده با جهش ژنی یکسان، تفاوتهای فردی زیادی در تظاهرات بالینی (بیان متغیر) دیده میشود1).
مطالعات کوهورت بزرگ نشان داده است که جهشهای FOXC1 و PITX2 با طیف وسیعی از گلوکوم از دوران کودکی تا بزرگسالی مرتبط هستند9). مواردی که در ابتدا به عنوان PCG (گلوکوم مادرزادی اولیه) تشخیص داده شدهاند، ممکن است پس از آزمایش ژنتیکی دوباره طبقهبندی شوند. در نوزادانی که یافتههای بخش قدامی چشم ظریف هستند، آزمایش ژنتیک به تشخیص دقیق نوع بیماری کمک میکند.
در مواردی که حذف کوچک در اطراف ژن PITX2 وجود دارد، حذف ژنهای NEUROG2، UGT8 و NDST4 ممکن است باعث تاخیر رشد و ناتوانی ذهنی شود8)3).
Kawanami و همکاران (2023) یک پسر ۳ ساله ژاپنی با حذف کوچک ۲.۵ مگابازی در ناحیه 4q25 (شامل PITX2، NEUROG2 و ANK2) گزارش کردند. او فتق نافی، کلوبوم عنبیه و تاخیر رشد داشت، اما نوار قلب با وجود حذف ANK2 طبیعی بود. هاپلوناکافی NEUROG2 به عنوان علت احتمالی تاخیر رشد در نظر گرفته شد8).
به دلیل وراثت اتوزومال غالب، احتمال انتقال جهش از والد مبتلا ۵۰٪ است. نفوذ کامل است اما فنوتیپ متغیر است و حتی با جهش یکسان، شدت علائم میتواند بسیار متفاوت باشد1). انجام آزمایش ژنتیک و مشاوره ژنتیک توصیه میشود.
تشخیص ARS بر اساس ناهنجاریهای دوطرفه زاویه و عنبیه است. چسبندگی جزئی عنبیه محیطی به خط شاگاس خلفی (posterior embryotoxon) شرط تشخیص در نظر گرفته میشود13). در مواردی که خط شاگاس خلفی با لامپ شکاف قابل مشاهده نیست، گونیوسکوپی ضروری است. در صورت وجود ناهنجاریهای سیستمیک، به عنوان سندرم ARS به متخصص اطفال برای بررسی کامل ارجاع داده میشود13).
توجه داشته باشید که ۸ تا ۱۵٪ از جمعیت طبیعی دارای خط شاگاس خلفی خفیف هستند، اما به تنهایی با گلوکوم همراه نیست. گرفتن سابقه خانوادگی نیز در تشخیص مهم است.
در سیستم طبقهبندی گلوکوم اطفال در راهنمای بالینی گلوکوم (ویرایش پنجم)، ARS به عنوان نمونه بارز گلوکوم مرتبط با ناهنجاریهای مادرزادی چشم در نظر گرفته میشود13). در صورت وجود ناهنجاری چشمی از بدو تولد که معیارهای تشخیص گلوکوم اطفال را داشته باشد، تشخیص داده میشود.
بیماریهای اصلی که باید از ARS افتراق داده شوند در زیر آورده شده است.
| بیماری | تفاوت با ARS |
|---|---|
| سندرم ICE | یک طرفه، اکتسابی، غالب در زنان |
| ناهنجاری پیترز | کدورت مرکزی قرنیه، فقدان غشای دسمه |
| آنیریدیا | پانوس قرنیه، هیپوپلازی فووئا |
| دیستروفی پلیمورف خلفی قرنیه | دو طرفه، خانوادگی، بدون تفاوت جنسی |
سندرم ICE (شامل آتروفی عنبیه پیشرونده و سندرم چندلر) نیاز به افتراق از ARS دارد، اما مهمترین نکته افتراقی این است که ICE یک طرفه و اکتسابی است در حالی که ARS دو طرفه و مادرزادی است.
در حال حاضر درمان قطعی برای خود ARS وجود ندارد و مدیریت گلوکوم و پایش عوارض سیستمیک محور اصلی درمان هستند. استراتژی درمان مشابه گلوکوم رشدی زودرس (گلوکوم مادرزادی اولیه: PCG) است13).
گلوکوم در حدود 50 تا 60 درصد موارد ARS همراه است. درمان دارویی مشابه گلوکوم عمومی انجام میشود، اما اغلب بیاثر است.
داروهای مهارکننده تولید زلالیه
مسدودکنندههای بتا: یکی از گزینههای خط اول. ایمن و مؤثر، اما در کودکان اغلب بیاثر است.
مهارکنندههای کربنیک آنهیدراز (CAI) موضعی: مانند برینزولامید. قابل استفاده همراه با مسدودکنندههای بتا.
آگونیستهای آلفا-2 (بریمونیدین): در کودکان زیر 2 سال به دلیل عوارض عصبی-روانی (آپنه، برادیکاردی، افت فشار خون، هیپوتونی عضلانی، افسردگی CNS) منع مصرف دارد13).
داروهای افزایش دهنده خروج زلالیه
داروهای مرتبط با پروستاگلاندین: مانند لاتانوپروست، تراووپروست. اثربخشی در کودکان نسبت به بزرگسالان ضعیفتر است13).
مثال بالینی: در یک پسر 7 ساله، تراووپروست + برینزولامید برای مدیریت طولانیمدت استفاده شد5). در یک مرد 77 ساله، لاتانوپروست/تیمولول + برینزولامید نیز IOP را 35 میلیمتر جیوه نگه داشت که کنترل دشوار بود2).
گزارش شده است که بین آگونیستهای گیرنده FP پروستانوئید و مسدودکنندههای بتا تفاوتی در اثربخشی وجود ندارد13).
در نوزادان و کودکان خردسال، به دلیل وزن و سطح بدن کوچک، دوز نسبی قطرههای چشمی بیشتر است، بنابراین باید تا حد امکان از داروهای با غلظت پایین استفاده کرد13).
اگر کنترل فشار چشم با دارو امکانپذیر نباشد، جراحی انجام میشود10)13).
عوارض پس از جراحی GDD شامل: اتاق قدامی کم عمق 13.6%، فشار خون پایین 11.7%، افیوژن مشیمیه 8.3% و اندوفتالمیت 1.7% گزارش شده است14).
Chakraborty و همکاران (2022) یک مورد از جداشدگی شبکیه همراه با ARS (پسر 15 ساله) را گزارش کردند. با میکروسفری و سابلوکساسیون عدسی همراه بود و پس از ویترکتومی، IOP به 41 میلیمتر جیوه افزایش یافت و استافیلوم تشکیل شد. سیکلو فوتوکواگولاسیون با دیود انجام شد و در نهایت IOP 18 میلیمتر جیوه به دست آمد7).
میزان موفقیت زاویهسازی (گونیوتومی) کمتر از PCG است 13). در ترابکولکتومی همراه با MMC، میزان موفقیت طولانی مدت ۲ ساله حدود ۵۹٪ و در دستگاههای تخلیه گلوکوم (GDD) در ۱۲ ماه ۸۷٪ و در ۲۴ ماه ۷۷٪ گزارش شده است 14). در موارد مقاوم، ممکن است نیاز به چندین بار جراحی باشد.
علت اصلی ARS نقص در مهاجرت و تمایز سلولهای تاج عصبی است. اختلال در رشد سلولهای تاج عصبی در اتاق قدامی، زاویه بخش قدامی چشم، استخوانهای صورت، دندانها، سیستم قلبی عروقی و پوست اطراف ناف منجر به ناهنجاریهای چندعضوی میشود.
در اواخر دوره جنینی، سلولهای اندوتلیال تمایز نیافته که اتاق قدامی را میپوشانند، به طور طبیعی از عنبیه و زاویه ناپدید میشوند. در ARS این روند ناپدید شدن مختل میشود و سلولهای اندوتلیال تمایز نیافته روی عنبیه باقی میمانند و باعث تشکیل رشتههای بافتی میشوند. در زاویه، اتصال بالای عنبیه رخ میدهد که به طور مکانیکی ترابکولوم را میپوشاند.
از نظر بافتشناسی، یک لایه منفرد از سلولهای شبه اندوتلیال همراه با غشایی شبه دسمه از سطح خلفی قرنیه تا اتاق قدامی، زاویه و سطح عنبیه به طور غیرطبیعی گسترش مییابد. این غشا در ربعهایی که اکتروپیون عنبیه و انحراف مردمک وجود دارد، دیده میشود و در ربع مقابل، آتروفی عنبیه مشاهده میشود.
FOXC1 و PITX2 هر دو فاکتورهای رونویسی هستند که به توالیهای خاص DNA متصل شده و بیان ژنهای پاییندست را تنظیم میکنند. این دو در تکامل بخش قدامی چشم به صورت هم افزایی عمل کرده و ژنهای هدف مشترک پاییندست را تنظیم میکنند 3). دامنه فورکهِد FOXC1 (یک دامنه اتصال به DNA با ۱۱۰ اسید آمینه) از نظر عملکردی مهمترین است 2) و جهش در این دامنه با علائم عصبی-روانی ارتباط قویتری دارد.
دو مکانیسم زیر برای افزایش فشار داخل چشم مطرح شده است:
میزان نقص عنبیه و میزان برآمدگیهای زاویهای عنبیه لزوماً با شدت گلوکوم همبستگی ندارد. با این حال، اگر چسبندگی عنبیه در زاویه شدید باشد، احتمال ابتلا به گلوکوم بیشتر میشود.
جهش FOXC1 در مقایسه با سایر جهشها، گلوکوم مادرزادی را بیشتر تسریع میکند 1) و ناهنجاریهای مورفولوژیک جسم مژگانی و زاویه تخلیه ممکن است در افزایش فشار داخل چشم نقش داشته باشند 1).
در موشهای دارای جهش FOXC1، کاهش و ناهنجاری ساختاری فیبرهای کلاژن در استرومای قرنیه و آسیب سلولهای استرومایی مشاهده میشود 1). علاوه بر این، FOXC1 به عنوان یک عامل مهارکننده رگزایی قرنیه عمل میکند (از طریق کنترل دسترسی زیستی VEGF) 1) و با از دست رفتن این مهار در اثر جهش FOXC1، رگزایی قرنیه رخ میدهد.
FOXC1 به عنوان یک فاکتور رونویسی از خانواده FOX، نقش مهمی در رشد مغز نیز ایفا میکند 4).
در یک مرور سیستماتیک، ناهنجاریهای ماده سفید در ۴۱.۳٪ از موارد ARS گزارش شده است 4). جهش FOXC1 میتواند باعث بیماری عروق کوچک مغزی، سیگنالهای بالای ماده سفید، فضاهای اطراف عروقی بزرگشده، میکروخونریزی و انفارکتوس لاکونار شود.
Ohkubo و همکاران (۲۰۲۵) در یک پسر ۲ ساله ژاپنی (جهش FOXC1: c.240del, p.Y81Ifs21) با MRI مغز، ضایعات ماده سفید اطراف بطنی، فضاهای اطراف عروقی بزرگشده و پیچخوردگی و گشادشدگی شریانهای مهرهای-قاعدهای را تأیید کردند. پدر وی سابقه سکته مغزی در سن ۱۸ سالگی داشت 4).
در مرور ۹۵ مورد جهش FOXC1، ۶.۳٪ علائم عصبی-روانی (مشکلات یادگیری، صرع، ناتوانی ذهنی، هذیان حسادت و غیره) داشتند و در ۸۳.۳٪ موارد با جهش در دامنه forkhead، علائم عصبی-روانی ظاهر شد 2).
بله. یک مرور سیستماتیک نشان میدهد که در ARS با جهش FOXC1، ناهنجاریهای ماده سفید در حدود ۴۱٪ موارد ظاهر میشود 4). جهش FOXC1 با خطر بیماری عروق کوچک مغزی و سکته مغزی مرتبط است و به ویژه در ARS ناشی از جهش FOXC1، پیگیری طولانیمدت عصبی مهم است.
در سالهای اخیر، با استفاده از توالییابی نسل جدید و توالییابی کامل ژنوم، جهشهای جدید زیادی گزارش شده است.
Wowra و همکاران (2024) در سه خواهر لهستانی مبتلا به ARS، یک حذف بزرگ (جهش جدید) شامل بخشی از اگزون 1 و کل ناحیه 3’UTR ژن FOXC1 شناسایی کردند. فنوتیپ در یک خانواده به طور قابل توجهی متفاوت بود و در ابتدا به اشتباه سندرم چندلر تشخیص داده شده بود1).
Jiang و همکاران (2024) در یک خانواده چینی مبتلا به ARS نوع 1، یک بازآرایی پیچیده ژنومی شامل حذف 6.15 مگابازی در کروموزوم 4q25 حاوی PITX2، وارونگی 45.71 مگابازی و حذف 14 جفت باز شناسایی کردند. یک دختر 11 ساله IOP 43.5/44.0 میلیمتر جیوه داشت3).
سایر گزارشهای جهش جدید شامل FOXC1 p.Phe136Leu (دامنه forkhead)2)، FOXC1 p.S82R (جهش de novo)5) و FOXC1 c.240del, p.Y81Ifs214) میباشد.
Yoshino و همکاران (2024) یک مورد ARS نوع 3 را در یک مرد 77 ساله ژاپنی گزارش کردند. از سن 72 سالگی هذیان حسادت ظاهر شد و لوکوانسفالوپاتی تأیید شد. در مرور 95 مورد جهش FOXC1، 6.3% (6/95) علائم عصبی-روانی داشتند که 83.3% (5/6) از آنها جهش در دامنه forkhead داشتند2).
این یافته نشان میدهد که دامنه عملکردی جهش FOXC1 ممکن است در بروز علائم عصبی-روانی نقش داشته باشد و اهمیت پیگیری طولانی مدت از نظر سلامت روان را نشان میدهد.
نشان داده شده است که جهش FOXC1 ممکن است باعث CSVD شده و خطر سکته مغزی را افزایش دهد4). پیشگیری و مداخله زودهنگام بیماریهای عصبی-عروقی در بیماران ARS به عنوان موضوع تحقیقات آینده مطرح است.
درک پاتوژنز «صلبیهای شدن» قرنیه ناشی از جهش FOXC1 در حال پیشرفت است1). انتظار میرود که درک مکانیسم مولکولی کدورت قرنیه منجر به توسعه درمانهای جدید با استفاده از ژن درمانی، داروهای ضد فیبروز و مواد زیستی شود1).