ARS tipe 1
Gen penyebab: PITX2 (4q25)
Kelainan utama: Kelainan segmen anterior mata, kelainan gigi, kulit berlebih di sekitar pusar/hernia umbilikalis, kelainan kraniofasial, kelainan kardiovaskular

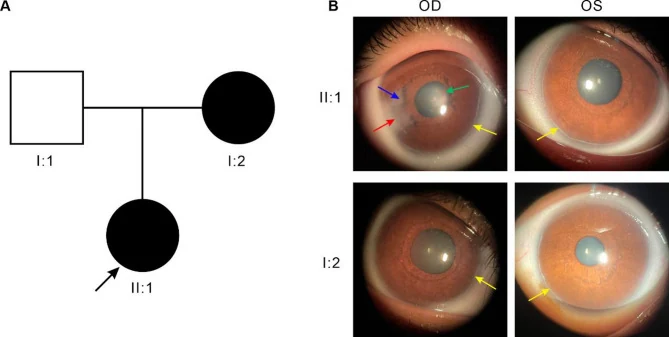
Sindrom Axenfeld-Rieger (ARS) adalah kelompok penyakit kongenital yang menggabungkan kelainan segmen anterior dan kelainan sistemik. Penyebab mendasar adalah gangguan migrasi dan diferensiasi sel krista neural. Pada akhir masa kehamilan, proses hilangnya sel endotel yang belum berdiferensiasi yang menutupi bilik mata depan dari iris dan sudut terganggu, dan sisa-sisanya menyebabkan pembentukan pita atau perlekatan iris yang tinggi.
Latar belakang sejarah: Pada tahun 1920, Axenfeld mendeskripsikan cincin Schwalbe posterior (pergeseran anterior dan penebalan garis Schwalbe) dan tonjolan iris. Pada tahun 1934-1935, Rieger menambahkan laporan tentang hipoplasia iris, deviasi pupil, dan polikoria. Saat ini diklasifikasikan dalam tiga tahap:
Semua ini secara kolektif disebut sindrom Axenfeld-Rieger. 50-60% disertai glaukoma, bersifat autosomal dominan dan biasanya bilateral. Sering juga disertai katarak dan ektopia lentis.
Epidemiologi: Prevalensi diperkirakan sekitar 1/200.000, namun laporan terbaru memperkirakan 1/50.000 hingga 1/100.0002)4). Tidak ada perbedaan jenis kelamin, dan sering didiagnosis pada masa bayi atau anak usia dini.
Klasifikasi genetik adalah sebagai berikut:
ARS tipe 1
Gen penyebab: PITX2 (4q25)
Kelainan utama: Kelainan segmen anterior mata, kelainan gigi, kulit berlebih di sekitar pusar/hernia umbilikalis, kelainan kraniofasial, kelainan kardiovaskular
ARS tipe 2
Gen penyebab: 13q14 (belum pasti)
Kelainan utama: Kelainan segmen anterior mata, glaukoma. Kelainan sistemik lebih sedikit dibandingkan tipe 1 dan 3
ARS tipe 3
Gen penyebab: FOXC1 (6p25)
Kelainan utama: Kelainan segmen anterior mata, glaukoma, gangguan pendengaran sensorineural, defek septum atrium, kelainan ginjal, lesi substansia alba
Mutasi FOXC1 dan PITX2 mencakup 40–70% kasus ARS5). Namun, pada 60% kasus ARS, gen penyebab belum teridentifikasi4), menunjukkan keragaman genetik yang besar.
Dalam analisis registri besar glaukoma onset anak dan dewasa muda, tingkat diagnosis molekuler adalah 56,5%11). Mutasi FOXC1 mencakup 20,3%, PITX2 17,4%, dan PAX6 10,1%, dan masih banyak kasus yang tidak dapat dijelaskan oleh gen yang diketahui11).
Mereka dibedakan berdasarkan gen penyebab. Tipe 1 disebabkan oleh mutasi PITX2 (4q25) dan disertai kelainan gigi, pusar, dan tulang wajah. Tipe 3 disebabkan oleh mutasi FOXC1 (6p25) dan disertai gangguan pendengaran, defek jantung, kelainan ginjal, dan kelainan neurologis. Tipe 2 terletak pada 13q14 tetapi gen penyebab belum pasti, dengan kelainan segmen anterior dan glaukoma sebagai ciri utama. Diagnosis dapat dipastikan dengan tes genetik.
Item utama temuan mata ditunjukkan di bawah ini.
| Temuan mata | Karakteristik |
|---|---|
| Cincin embrionik posterior | Pergeseran anterior dan penebalan garis Schwalbe |
| Proyeksi iris | Berbentuk filamen halus hingga pita lebar |
| Deviasi pupil | Deviasi ke arah berlawanan dari anulus embrional posterior |
| Pseudopolikoria | Penampilan seperti perforasi pada stroma iris |
| Eversi uvea | Eversi epitel pigmen iris |
Anulus embrional posterior adalah sisa sel yang tidak berdiferensiasi di garis Schwalbe, tampak linear sepanjang limbus pada 0,5–2,0 mm ke arah sentral dari limbus. Seringkali hanya terbatas pada sebagian area, tidak melingkar penuh. Jika garis Schwalbe yang menonjol melekat pada iris, disebut anomali Axenfeld; jika disertai atrofi stroma iris, disebut anomali Rieger.
Kornea biasanya jernih dengan struktur endotel normal, namun kontak fisik dengan jaringan sisa dapat menyebabkan kekeruhan kornea sekunder. Kekeruhan sering terbatas pada bagian perifer dan umumnya tidak langsung mempengaruhi penglihatan. Namun, pada mutasi FOXC1, kekeruhan dan neovaskularisasi kornea lebih menonjol, dan tingkat kelainan kornea serta frekuensi glaukoma lebih tinggi dibandingkan mutasi PITX21).
Temuan sudut meliputi perlekatan iris yang tinggi, sisa uvea seperti tali, dan penebalan garis Schwalbe (anulus embrional posterior). Dilaporkan juga kasus dengan lensa mikrosferofakia atau subluksasi lensa7).
Glaukoma terjadi pada 50–60% kasus. Peningkatan tekanan intraokular dapat terjadi sejak bayi, namun sebagian besar muncul pada masa kanak-kanak hingga dewasa muda. Beberapa kasus didiagnosis setelah penurunan penglihatan progresif, sehingga penting untuk tidak melewatkan temuan segmen anterior dan perubahan glaukoma6).
Pada kasus Li et al. (2021) seorang anak laki-laki 7 tahun (ARS tipe 3, mutasi de novo FOXC1), diameter kornea 14 mm, panjang aksial 27,16/26,56 mm, rasio C/D 0,9, TIO 33/20 mmHg. Kedua mata memerlukan operasi antiglaukoma sejak hari ke-36 setelah lahir5).
Temuan sistemik adalah sebagai berikut:
Sekitar 50-60% pasien ARS mengalami glaukoma. Sering muncul pada masa kanak-kanak hingga dewasa muda, tetapi ada juga kasus dengan peningkatan tekanan intraokular sejak bayi. Diperlukan pengukuran tekanan intraokular secara teratur dan evaluasi saraf optik. Untuk detailnya, lihat bagian “Metode pengobatan standar”.
ARS menunjukkan pola pewarisan autosomal dominan dengan penetrasi penuh. Namun, meskipun dalam keluarga yang sama dengan mutasi gen yang sama, terdapat variasi klinis yang besar (variable expressivity)1).
Dalam studi kohort besar, mutasi FOXC1 dan PITX2 terkait dengan spektrum glaukoma yang luas dari masa kanak-kanak hingga dewasa9). Kasus yang awalnya didiagnosis secara klinis sebagai glaukoma kongenital primer (PCG) kadang direklasifikasi melalui tes genetik, dan tes genetik berkontribusi pada diagnosis tipe yang akurat ketika temuan segmen anterior pada bayi tidak jelas.
Pada kasus dengan mikrodelesi di sekitar gen PITX2, tumpang tindih delesi NEUROG2, UGT8, dan NDST4 dapat menyebabkan keterlambatan perkembangan dan disabilitas intelektual 8)3).
Kawanami et al. (2023) melaporkan seorang anak laki-laki Jepang berusia 3 tahun dengan mikrodelesi 2,5 Mb pada 4q25 (termasuk PITX2, NEUROG2, dan ANK2). Ia mengalami hernia umbilikalis, koloboma iris, dan keterlambatan perkembangan, namun EKG normal meskipun ada delesi ANK2. Haploinsufisiensi NEUROG2 dianggap sebagai kandidat penyebab keterlambatan perkembangan 8).
Karena bersifat autosomal dominan, probabilitas pewarisan mutasi dari orang tua adalah 50%. Penetrasi lengkap tetapi fenotipe bervariasi, dan tingkat keparahan gejala dapat sangat berbeda meskipun dengan mutasi yang sama 1). Pemeriksaan genetik dan konseling genetik direkomendasikan.
Diagnosis ARS didasarkan pada kelainan sudut bilik mata depan dan iris pada kedua mata. Adanya posterior embryotoxon dengan perlekatan sebagian iris di sekitarnya merupakan syarat diagnosis 13). Jika posterior embryotoxon tidak terlihat dengan slit lamp, diperlukan gonioskopi. Jika terdapat kelainan sistemik, pasien dirujuk ke dokter anak untuk evaluasi menyeluruh sebagai bagian dari sindrom ARS 13).
Perlu dicatat bahwa 8-15% populasi normal memiliki posterior embryotoxon ringan, tetapi ini saja tidak disertai glaukoma atau lainnya. Riwayat keluarga juga penting dalam diagnosis.
Dalam sistem klasifikasi glaukoma pediatrik menurut Pedoman Penanganan Glaukoma (Edisi ke-5), ARS diklasifikasikan sebagai contoh tipikal glaukoma terkait kelainan perkembangan okular kongenital 13). Diagnosis ditegakkan ketika kelainan okular yang ada sejak lahir memenuhi kriteria diagnosis glaukoma pediatrik.
Berikut adalah penyakit utama yang harus dibedakan dari ARS.
| Penyakit | Perbedaan dari ARS |
|---|---|
| Sindrom ICE | Unilateral, didapat, dominan wanita |
| Anomali Peters | Kekeruhan kornea sentral, defek membran Descemet |
| Aniridia | Pannus kornea, hipoplasia fovea |
| Distrofi kornea polimorf posterior | Bilateral, familial, tidak ada perbedaan jenis kelamin |
Sindrom ICE (seperti atrofi iris progresif, sindrom Chandler) penting untuk dibedakan dari ARS, tetapi perbedaan utamanya adalah ICE bersifat unilateral dan didapat, sedangkan ARS bersifat bilateral dan kongenital.
Saat ini tidak ada terapi kuratif untuk ARS itu sendiri, dan pengelolaan glaukoma serta surveilans komplikasi sistemik menjadi inti pengobatan. Pendekatan pengobatan mengikuti glaukoma perkembangan awal (glaukoma kongenital primer: PCG) 13).
Glaukoma terjadi pada sekitar 50-60% kasus ARS. Terapi obat dilakukan sesuai dengan glaukoma umum, tetapi seringkali tidak efektif.
Obat Penekan Produksi Humor Akuos
Beta-blocker: Salah satu pilihan pertama. Aman dan efektif tetapi seringkali tidak efektif pada anak-anak.
Tetes penghambat karbonat anhidrase (CAI): seperti brinzolamid. Dapat dikombinasikan dengan beta-blocker.
Agonis alfa-2 (brimonidin): Kontraindikasi pada anak di bawah 2 tahun karena efek neuropsikiatri (apnea, bradikardia, hipotensi, hipotonia, depresi SSP) 13).
Obat Peningkat Aliran Humor Akuos
Analog prostaglandin: seperti latanoprost, travoprost. Efeknya pada anak-anak lebih lemah dibandingkan dewasa 13).
Contoh: Pada kasus anak laki-laki 7 tahun, pengelolaan jangka panjang dengan travoprost + brinzolamid 5). Pada kasus pria 77 tahun, TIO 35 mmHg meskipun dengan latanoprost/timolol + brinzolamid, menunjukkan kontrol sulit 2).
Ada laporan bahwa tidak ada perbedaan efektivitas antara agonis reseptor FP prostanoid dan beta-blocker 13).
Pada bayi dan anak kecil, dosis tetes mata relatif besar dibandingkan berat badan dan luas permukaan tubuh, sehingga sebaiknya gunakan konsentrasi obat serendah mungkin 13).
Jika tekanan intraokular tidak terkontrol dengan obat, dilakukan pembedahan10)13).
Komplikasi pasca GDD meliputi: bilik mata depan dangkal 13,6%, hipotoni 11,7%, efusi koroid 8,3%, dan endoftalmitis 1,7%14).
Chakraborty dkk. (2022) melaporkan satu kasus ablasi retina terkait ARS (anak laki-laki 15 tahun). Disertai mikrofakia dan subluksasi lensa, setelah vitrektomi tekanan intraokular meningkat menjadi 41 mmHg dan terbentuk stafiloma. Dilakukan fotokoagulasi badan siliaris dioda dan akhirnya tekanan intraokular mencapai 18 mmHg7).
Tingkat keberhasilan operasi sudut lebih rendah dibandingkan PCG 13). Pada trabekulektomi dengan MMC, tingkat keberhasilan jangka panjang 2 tahun sekitar 59%, sedangkan pada GDD dilaporkan 87% pada 12 bulan dan 77% pada 24 bulan 14). Kasus refrakter mungkin memerlukan beberapa kali operasi.
Penyebab mendasar ARS adalah cacat migrasi dan diferensiasi sel krista neural. Gangguan perkembangan sel krista neural di bilik mata depan, sudut mata anterior, tulang wajah, gigi, sistem kardiovaskular, dan kulit peri-umbilikal menyebabkan malformasi multi-organ.
Pada akhir masa kehamilan, sel endotel yang belum berdiferensiasi yang menutupi bilik mata depan biasanya menghilang dari iris dan sudut. Pada ARS, proses penghilangan ini terganggu, dan sel endotel yang belum berdiferensiasi menetap di iris, menyebabkan pembentukan pita. Di sudut, terjadi perlekatan iris yang tinggi, yang secara mekanis menutupi trabekula.
Secara histologis, terdapat lapisan tunggal sel seperti endotel dengan membran seperti membran Descemet yang meluas secara abnormal dari permukaan posterior kornea melalui bilik mata depan, sudut, dan permukaan iris. Membran terdapat di kuadran dengan eversi uvea dan deviasi pupil, sedangkan atrofi iris terlihat di kuadran yang berlawanan.
FOXC1 dan PITX2 keduanya adalah faktor transkripsi, yang mengikat sekuens DNA spesifik dan mengatur ekspresi gen hilir. Keduanya bekerja secara sinergis dalam perkembangan segmen anterior mata dan mengatur gen target hilir yang sama 3). Domain forkhead FOXC1 (domain pengikat DNA 110 asam amino) adalah yang paling penting secara fungsional 2), dan mutasi pada domain ini diduga terkait lebih kuat dengan gejala neuropsikiatri.
Dua mekanisme peningkatan tekanan intraokular telah diidentifikasi:
Tingkat defek iris dan jumlah tonjolan iris sudut tidak selalu berkorelasi dengan keparahan glaukoma. Namun, perlengketan iris sudut yang parah meningkatkan risiko glaukoma.
Mutasi FOXC1 lebih mendorong glaukoma kongenital dibandingkan mutasi lainnya 1), dan kelainan morfologi badan siliaris serta sudut drainase mungkin berperan dalam peningkatan TIO 1).
Pada tikus dengan mutasi FOXC1, diamati penurunan serat kolagen stroma kornea, kelainan struktural, dan kerusakan sel stroma 1). Selain itu, FOXC1 berfungsi sebagai faktor penekan neovaskularisasi kornea (melalui regulasi bioavailabilitas VEGF) 1), dan hilangnya penekanan ini akibat mutasi FOXC1 menyebabkan neovaskularisasi kornea.
FOXC1, sebagai faktor transkripsi keluarga FOX, juga memainkan peran penting dalam perkembangan otak 4).
Dalam tinjauan sistematis, dilaporkan bahwa kelainan materi putih muncul pada 41,3% kasus ARS 4). Mutasi FOXC1 dapat menginduksi penyakit pembuluh darah kecil otak, hiperintensitas materi putih, perluasan ruang perivaskular, mikroperdarahan, dan infark lakunar.
Ohkubo dkk. (2025) mengonfirmasi lesi materi putih periventrikular, perluasan ruang perivaskular, serta tortuositas dan ektasia arteri vertebrobasilar pada MRI otak seorang anak laki-laki Jepang berusia 2 tahun (mutasi FOXC1: c.240del, p.Y81Ifs21). Ayahnya memiliki riwayat infark serebral pada usia 18 tahun 4).
Dalam tinjauan 95 kasus mutasi FOXC1, gejala neuropsikiatri (kesulitan belajar, epilepsi, disabilitas intelektual, delusi kecemburuan, dll.) ditemukan pada 6,3%, dan gejala neuropsikiatri muncul pada 83,3% kasus dengan mutasi domain forkhead 2).
Ya. Ada tinjauan sistematis yang melaporkan bahwa kelainan materi putih muncul pada sekitar 41% kasus ARS dengan mutasi FOXC1 4). Mutasi FOXC1 diduga terkait dengan penyakit pembuluh darah kecil otak dan risiko stroke, terutama pada ARS tipe FOXC1, pemantauan neurologis jangka panjang penting dilakukan.
Dalam beberapa tahun terakhir, banyak mutasi baru telah dilaporkan melalui sekuensing generasi berikutnya dan sekuensing seluruh genom.
Wowra dkk. (2024) mengidentifikasi delesi besar (mutasi baru) yang mencakup sebagian ekson 1 dan seluruh 3’UTR dari FOXC1 pada tiga saudara perempuan Polandia dengan ARS. Fenotipe sangat bervariasi bahkan dalam keluarga yang sama, dan awalnya salah didiagnosis sebagai sindrom Chandler 1).
Jiang dkk. (2024) mengidentifikasi penataan ulang genom kompleks yang terdiri dari delesi 6,15 Mb pada kromosom 4q25 yang mencakup PITX2, inversi 45,71 Mb, dan delesi 14 bp pada sebuah keluarga Tiongkok dengan ARS tipe 1. Seorang gadis berusia 11 tahun menunjukkan IOP 43,5/44,0 mmHg 3).
Laporan mutasi baru lainnya termasuk FOXC1 p.Phe136Leu (domain forkhead) 2), FOXC1 p.S82R (mutasi de novo) 5), dan FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino dkk. (2024) melaporkan kasus seorang pria Jepang berusia 77 tahun dengan ARS tipe 3. Delusi kecemburuan muncul sejak usia 72 tahun, dan leukoensefalopati dikonfirmasi. Dalam tinjauan literatur terhadap 95 kasus mutasi FOXC1, gejala neuropsikiatri ditemukan pada 6,3% (6/95), dan 83,3% (5/6) di antaranya memiliki mutasi domain forkhead 2).
Temuan ini menunjukkan bahwa domain fungsional mutasi FOXC1 mungkin terlibat dalam manifestasi gejala neuropsikiatri, menyoroti pentingnya tindak lanjut jangka panjang dari perspektif kesehatan mental.
Mutasi FOXC1 telah diindikasikan dapat menginduksi CSVD dan meningkatkan risiko stroke 4). Pencegahan dan intervensi dini penyakit neurovaskular pada pasien ARS merupakan topik penelitian masa depan.
Pemahaman tentang patogenesis “skleralisasi” kornea akibat mutasi FOXC1 sedang berkembang 1). Pemahaman mekanisme molekuler kekeruhan kornea diharapkan dapat mengarah pada pengembangan terapi baru menggunakan terapi gen, obat antifibrotik, dan biomaterial 1).