ARS type 1
Causative gene: PITX2 (4q25)
Main features: Anterior segment anomalies, dental anomalies, redundant periumbilical skin/umbilical hernia, craniofacial anomalies, cardiovascular anomalies

Axenfeld-Rieger syndrome (ARS) is a group of congenital disorders combining anterior segment dysgenesis and systemic abnormalities. The fundamental etiology is thought to be abnormal neural crest cell migration and differentiation. The normal process of disappearance of undifferentiated endothelial cells covering the anterior chamber from the iris and angle in late fetal life is impaired, and their remnants cause strand formation and high iris insertion.
Historical background: In 1920, Axenfeld described posterior embryotoxon (anterior displacement and thickening of Schwalbe’s line) and iris processes. In 1934-1935, Rieger added reports of iris hypoplasia, corectopia, and polycoria. Currently, it is classified into the following three stages:
These are collectively called Axenfeld-Rieger syndrome. Glaucoma occurs in 50-60% of cases, and it is usually bilateral with autosomal dominant inheritance. Cataract and lens dislocation are also frequently associated.
Epidemiology: The prevalence has been reported as approximately 1 in 200,000, but recent estimates suggest 1 in 50,000 to 100,000 2)4). There is no sex predilection, and it is often diagnosed in infancy or early childhood.
Genetic classification is as follows:
ARS type 1
Causative gene: PITX2 (4q25)
Main features: Anterior segment anomalies, dental anomalies, redundant periumbilical skin/umbilical hernia, craniofacial anomalies, cardiovascular anomalies
ARS type 2
Causative gene: 13q14 (not yet identified)
Main features: Anterior segment anomalies, glaucoma. Systemic anomalies are less frequent than in types 1 and 3
ARS type 3
Causative gene: FOXC1 (6p25)
Main features: Anterior segment anomalies, glaucoma, sensorineural hearing loss, atrial septal defect, renal anomalies, white matter lesions
Mutations in FOXC1 and PITX2 account for 40–70% of ARS cases 5). However, the causative gene remains unidentified in 60% of ARS cases 4), indicating substantial genetic heterogeneity.
In a large registry analysis of childhood- and young adult-onset glaucoma, the molecular diagnostic rate was 56.5% 11). FOXC1 mutations accounted for 20.3%, PITX2 mutations for 17.4%, and PAX6 mutations for 10.1%; a considerable number of cases could not be explained by known genes 11).
They are distinguished by the causative gene. Type 1 is caused by PITX2 (4q25) mutations and is associated with dental, umbilical, and facial bone anomalies. Type 3 is caused by FOXC1 (6p25) mutations and is associated with hearing loss, heart defects, renal anomalies, and neurological abnormalities. Type 2 maps to 13q14 but the causative gene is not yet identified; it primarily presents with anterior segment anomalies and glaucoma. Genetic testing can provide a definitive diagnosis.
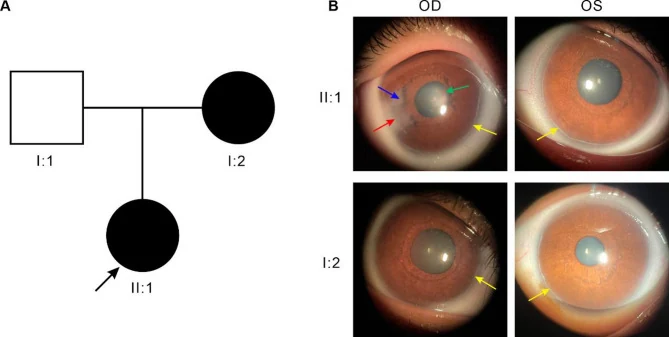
Major ocular findings are listed below.
| Ocular Finding | Characteristics |
|---|---|
| Posterior embryotoxon | Anterior displacement and thickening of Schwalbe’s line |
| Iris processes | Fine thread-like to broad band-like |
| Pupil deviation | Deviation opposite to posterior embryotoxon |
| Pseudopolycoria | Perforated appearance of iris stroma |
| Uveal ectropion | Eversion of iris pigment epithelium |
Posterior embryotoxon is a remnant of undifferentiated cells at the Schwalbe line, observed linearly along the limbus 0.5–2.0 mm centrally from the limbus. It is often localized rather than circumferential. Adhesion between the prominent Schwalbe line and the iris is called Axenfeld anomaly, and if accompanied by iris stromal atrophy, it is called Rieger anomaly.
The cornea is usually transparent with normal endothelial structure, but secondary corneal opacity may occur due to physical contact with residual tissue. Corneal opacity is often localized to the periphery and generally does not directly affect vision. However, in FOXC1 mutations, corneal opacity and corneal neovascularization are more prominent, and the degree of corneal abnormality is greater and the frequency of glaucoma is higher compared to PITX2 mutations 1).
Gonioscopic findings include high iris insertion, persistent pupillary membrane strands, and thickened Schwalbe line (posterior embryotoxon). Cases complicated by spherophakia or lens subluxation have also been reported 7).
Glaucoma occurs in 50–60% of cases. Intraocular pressure elevation may occur in infancy, but most cases develop in childhood to young adulthood. Some cases are diagnosed after progressive vision loss, so it is important not to overlook anterior segment findings and glaucomatous changes 6).
Li et al. (2021) reported a 7-year-old boy (ARS type 3, de novo FOXC1 mutation) with corneal diameter 14 mm, axial length 27.16/26.56 mm, cup-to-disc ratio 0.9, and IOP 33/20 mmHg. Antiglaucoma surgery was required for both eyes from 36 days after birth 5).
Systemic findings are as follows:
Glaucoma occurs in about 50–60% of ARS cases. It often develops during childhood to young adulthood, but some cases present with elevated intraocular pressure in infancy. Regular intraocular pressure measurement and optic nerve evaluation are necessary. For details, see the “Standard Treatment” section.
ARS shows autosomal dominant inheritance with complete penetrance. However, even within the same family carrying the same gene mutation, there is significant variability in clinical presentation (variable expressivity)1).
Large cohort studies have shown that FOXC1 and PITX2 mutations are associated with a broad spectrum of glaucoma from childhood to adulthood9). Cases initially diagnosed as PCG (primary congenital glaucoma) may be reclassified by genetic testing, and genetic testing contributes to accurate diagnosis when anterior segment findings in infants are subtle.
In cases with microdeletions around the PITX2 gene, overlapping deletions of NEUROG2, UGT8, and NDST4 can lead to developmental delay and intellectual disability 8)3).
Kawanami et al. (2023) reported a 3-year-old Japanese boy with a 2.5 Mb microdeletion at 4q25 (including PITX2, NEUROG2, and ANK2). He presented with omphalocele, iris coloboma, and developmental delay, but his electrocardiogram was normal despite the ANK2 deletion. Haploinsufficiency of NEUROG2 was considered a candidate cause of the developmental delay 8).
Due to autosomal dominant inheritance, the probability of transmission from an affected parent is 50%. Penetrance is complete, but the phenotype varies; even with the same mutation, symptom severity can differ greatly 1). Genetic testing and genetic counseling are recommended.
The diagnosis of ARS is based on bilateral angle abnormalities and iris abnormalities. Partial attachment of the peripheral iris to the posterior embryotoxon is considered a diagnostic criterion 13). If posterior embryotoxon is not visible on slit-lamp examination, gonioscopy is necessary. If systemic abnormalities are present, referral to pediatrics for a full systemic workup is indicated as ARS syndrome 13).
Note that a mild posterior embryotoxon is seen in 8–15% of the normal population, but alone it is not associated with glaucoma. Family history is also important in diagnosis.
In the classification system of childhood glaucoma in the Glaucoma Clinical Practice Guidelines (5th edition), ARS is positioned as a representative example of glaucoma associated with congenital ocular anomalies 13). It is diagnosed when ocular anomalies present at birth meet the diagnostic criteria for childhood glaucoma.
The main diseases to be differentiated from ARS are listed below.
| Disease | Differences from ARS |
|---|---|
| ICE syndrome | Unilateral, acquired, female predominance |
| Peters anomaly | Central corneal opacity, Descemet membrane defect |
| Aniridia | Corneal pannus, foveal hypoplasia |
| Posterior polymorphous corneal dystrophy | Bilateral, familial, no sex difference |
Differentiation from ICE syndrome (progressive iris atrophy, Chandler syndrome, etc.) is important, but the key distinguishing point is that ICE is unilateral and acquired, whereas ARS is bilateral and congenital.
There is currently no curative treatment for ARS itself; management focuses on glaucoma control and surveillance for systemic complications. Treatment strategy follows that of early-onset developmental glaucoma (primary congenital glaucoma: PCG) 13).
Glaucoma occurs in approximately 50–60% of ARS cases. Medication follows general glaucoma treatment but is often ineffective.
Aqueous Humor Suppressants
Beta-blockers: One of the first-line options. Safe and effective, but often ineffective in children.
Carbonic anhydrase inhibitors (CAI) eye drops: e.g., brinzolamide. Can be used in combination with beta-blockers.
Alpha-2 agonists (brimonidine): Contraindicated in children under 2 years due to neuropsychiatric side effects (apnea, bradycardia, hypotension, muscle hypotonia, central nervous system depression) 13).
Aqueous Humor Outflow Enhancers
Prostaglandin analogs: e.g., latanoprost, travoprost. Efficacy in children is considered weaker than in adults 13).
Example: A 7-year-old boy was managed long-term with travoprost + brinzolamide 5). A 77-year-old man had IOP of 35 mmHg despite latanoprost, timolol, and brinzolamide, indicating poor control 2).
There is a report that no difference in efficacy was found between prostanoid FP receptor agonists and beta-blockers 13).
In infants, the dose of eye drops is relatively high compared to body weight and body surface area; therefore, the lowest concentration possible should be used 13).
If intraocular pressure cannot be controlled with medication, surgery is performed10)13).
Complications after GDD surgery include shallow anterior chamber 13.6%, hypotony 11.7%, choroidal effusion 8.3%, and endophthalmitis 1.7%14).
Chakraborty et al. (2022) reported a case of ARS-associated retinal detachment (15-year-old boy). The patient had microspherophakia and lens subluxation; after vitrectomy, IOP rose to 41 mmHg and a scleral staphyloma formed. Diode cyclophotocoagulation was performed, ultimately achieving IOP of 18 mmHg7).
The success rate of angle surgery is lower than that for PCG13). For trabeculectomy with MMC, the 2-year long-term success rate is approximately 59%; for GDD, it is reported as 87% at 12 months and 77% at 24 months14). Refractory cases may require multiple surgeries.
The fundamental etiology of ARS is a defect in neural crest cell migration and differentiation. Impaired development of neural crest cells in the anterior chamber, anterior chamber angle, facial bones, teeth, cardiovascular system, and periumbilical skin leads to multi-organ malformations.
In the late embryonic stage, the undifferentiated endothelial cells covering the anterior chamber normally disappear from the iris and angle. In ARS, this disappearance process is impaired, and undifferentiated endothelial cells remain on the iris, causing strand formation. In the angle, high iris insertion occurs, mechanically covering the trabecular meshwork.
Histologically, a monolayer of endothelial-like cells with a Descemet-like membrane abnormally extends from the posterior cornea to the anterior chamber, angle, and iris surface. The membrane is present in quadrants with uveal ectropion and corectopia, while iris atrophy is observed in the opposite quadrants.
FOXC1 and PITX2 are both transcription factors that bind to specific DNA sequences and regulate the expression of downstream genes. They act synergistically in anterior segment development and regulate common downstream target genes3). The forkhead domain (110-amino acid DNA-binding domain) of FOXC1 is functionally most important2), and mutations in this domain are suggested to be more strongly associated with neuropsychiatric symptoms.
The following two mechanisms have been pointed out for intraocular pressure elevation.
The degree of iris defect and the amount of iris processes in the angle do not necessarily correlate with glaucoma severity. However, extensive peripheral anterior synechiae in the angle predispose to glaucoma.
FOXC1 mutations promote congenital glaucoma more than other mutations 1), and morphological abnormalities of the ciliary body and drainage angle may contribute to IOP elevation 1).
In FOXC1 mutant mice, reduced collagen fibers and structural abnormalities in the corneal stroma, as well as damage to keratocytes, are observed 1). Furthermore, FOXC1 functions as a suppressor of corneal angiogenesis (via regulation of VEGF bioavailability) 1), and loss of this suppression due to FOXC1 mutation leads to corneal neovascularization.
FOXC1, as a FOX family transcription factor, also plays an important role in brain development 4).
A systematic review reported that white matter abnormalities appear in 41.3% of ARS cases 4). FOXC1 mutations can induce cerebral small vessel disease (CSVD), white matter hyperintensities, enlarged perivascular spaces, microbleeds, and lacunar infarcts.
Ohkubo et al. (2025) confirmed periventricular white matter lesions, enlarged perivascular spaces, and vertebrobasilar dolichoectasia on brain MRI in a 2-year-old Japanese boy (FOXC1 mutation: c.240del, p.Y81Ifs21). His father had a history of cerebral infarction at age 18 4).
In a review of 95 FOXC1 mutation cases, 6.3% had neuropsychiatric symptoms (learning difficulties, epilepsy, intellectual disability, delusional jealousy, etc.), and 83.3% of cases with forkhead domain mutations exhibited neuropsychiatric symptoms 2).
Yes. A systematic review reported that white matter abnormalities appear in approximately 41% of ARS cases with FOXC1 mutations 4). FOXC1 mutations have been suggested to be associated with cerebral small vessel disease and stroke risk, and long-term neurological follow-up is particularly important in FOXC1-mutated ARS.
In recent years, many novel mutations have been reported through next-generation sequencing and whole-genome sequencing.
Wowra et al. (2024) identified a large deletion (novel mutation) involving part of FOXC1 exon 1 and the entire 3’UTR in three Polish sisters with ARS. Even within the same family, phenotypes varied greatly, and they were initially misdiagnosed with Chandler syndrome 1).
Jiang et al. (2024) identified a complex genomic rearrangement in a Chinese family with ARS type 1, including a 6.15 Mb deletion of chromosome 4q25 containing PITX2, a 45.71 Mb inversion, and a 14 bp deletion. An 11-year-old girl presented with IOP of 43.5/44.0 mmHg 3).
Other reports of novel mutations include FOXC1 p.Phe136Leu (forkhead domain) 2), FOXC1 p.S82R (de novo mutation) 5), and FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino et al. (2024) reported a case of ARS type 3 in a 77-year-old Japanese man. He developed delusions of jealousy at age 72, and leukoencephalopathy was confirmed. In a literature review of 95 FOXC1 mutation cases, neuropsychiatric symptoms were observed in 6.3% (6/95), and 83.3% (5/6) of those had forkhead domain mutations 2).
This finding suggests that the functional domain of FOXC1 mutations may be involved in the development of neuropsychiatric symptoms, highlighting the importance of long-term follow-up from a mental health perspective.
It has been suggested that FOXC1 mutations may induce CSVD and increase stroke risk 4). The importance of prevention and early intervention for neurovascular diseases in ARS patients is a future research topic.
The pathogenesis of corneal “sclerization” due to FOXC1 mutations is being elucidated 1). Understanding the molecular mechanisms of corneal opacity is expected to lead to the development of novel therapies using gene therapy, anti-fibrotic drugs, and biomaterials 1).