ARS tipo 1
Gene causale: PITX2 (4q25)
Anomalie principali: anomalie del segmento anteriore dell’occhio, anomalie dentali, eccesso di pelle periombelicale / ernia ombelicale, anomalie craniofacciali, anomalie cardiovascolari

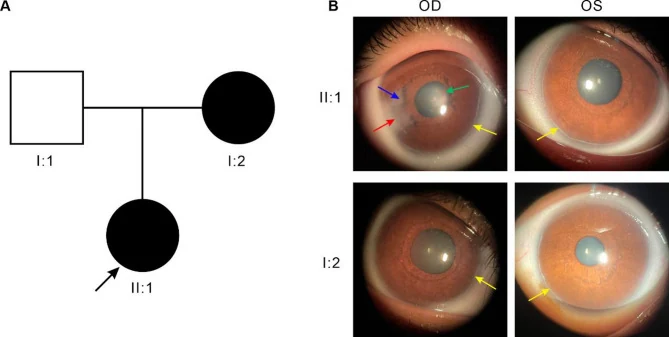
La sindrome di Axenfeld-Rieger (ARS) è un gruppo di malattie congenite che combinano anomalie del segmento anteriore dell’occhio e anomalie sistemiche. La causa di base è un’anomalia della migrazione e differenziazione delle cellule della cresta neurale. Alla fine del periodo fetale, il normale processo di scomparsa delle cellule endoteliali indifferenziate che rivestono la camera anteriore dall’iride e dall’angolo è alterato; la loro persistenza porta alla formazione di briglie e a un inserimento alto dell’iride.
Contesto storico: Nel 1920 Axenfeld descrisse l’anello embrionale posteriore (spostamento anteriore e ispessimento della linea di Schwalbe) e i processi iridei. Nel 1934-1935 Rieger aggiunse l’ipoplasia dell’iride, la deviazione pupillare e la policoria. Attualmente si classificano tre stadi:
Tutto ciò viene complessivamente chiamato sindrome di Axenfeld-Rieger. Nel 50-60% dei casi si sviluppa glaucoma, l’ereditarietà è autosomica dominante e di solito bilaterale. Cataratta ed ectopia lenticolare sono anch’esse frequentemente associate.
Epidemiologia: la prevalenza era stimata in circa 1/200.000, ma recenti rapporti la stimano tra 1/50.000 e 1/100.0002)4). Non c’è differenza di sesso e la diagnosi viene spesso posta nella prima infanzia.
Classificazione genetica:
ARS tipo 1
Gene causale: PITX2 (4q25)
Anomalie principali: anomalie del segmento anteriore dell’occhio, anomalie dentali, eccesso di pelle periombelicale / ernia ombelicale, anomalie craniofacciali, anomalie cardiovascolari
ARS tipo 2
Gene causale: 13q14 (non confermato)
Anomalie principali: anomalie del segmento anteriore dell’occhio, glaucoma. Le anomalie sistemiche sono meno frequenti rispetto ai tipi 1 e 3.
ARS tipo 3
Gene causale: FOXC1 (6p25)
Anomalie principali: anomalie del segmento anteriore dell’occhio, glaucoma, ipoacusia neurosensoriale, difetto del setto interatriale, anomalie renali, lesioni della sostanza bianca
Le mutazioni di FOXC1 e PITX2 rappresentano il 40-70% dei casi di ARS5). Tuttavia, nel 60% dei casi di ARS il gene causale non è identificato4), indicando una grande diversità genetica.
Un’analisi di un ampio registro di glaucoma infantile e giovanile ha mostrato un tasso di diagnosi molecolare del 56,5%11). Le mutazioni FOXC1 rappresentavano il 20,3%, PITX2 il 17,4% e PAX6 il 10,1%, e non pochi casi non possono essere spiegati dai geni noti11).
Si distinguono in base al gene causale. Il tipo 1 è causato da una mutazione di PITX2 (4q25) e si accompagna ad anomalie dentali, ombelicali e delle ossa facciali. Il tipo 3 è causato da una mutazione di FOXC1 (6p25) e si accompagna a sordità, difetti cardiaci, anomalie renali e neurologiche. Il tipo 2 è localizzato sul 13q14 ma il gene causale non è confermato; è caratterizzato principalmente da anomalie del segmento anteriore e glaucoma. La diagnosi può essere confermata tramite test genetico.
I principali segni oculari sono elencati di seguito.
| Segno oculare | Caratteristiche |
|---|---|
| Anello embrionale posteriore | Dislocazione anteriore e ispessimento della linea di Schwalbe |
| Processi iridei | Da filiformi a larghe bande |
| Deviazione pupillare | Deviazione nella direzione opposta all’anello embrionale posteriore |
| Pseudopolicoria | Aspetto perforato dello stroma irideo |
| Eversione uveale | Rovesciamento dell’epitelio pigmentato dell’iride |
L’anello embrionale posteriore è un residuo di cellule indifferenziate a livello della linea di Schwalbe, osservato come una linea lungo il limbo, spostata di 0,5–2,0 mm verso il centro. Spesso non è circonferenziale ma limitato a una parte. Se la linea di Schwalbe prominente aderisce all’iride, si parla di anomalia di Axenfeld; se è accompagnata da atrofia dello stroma irideo, si parla di anomalia di Rieger.
La cornea è solitamente trasparente e l’endotelio è normale, ma il contatto fisico con il tessuto residuo può causare secondariamente un’opacità corneale. L’opacità è spesso limitata alla periferia e generalmente non influisce direttamente sulla vista. Tuttavia, nelle mutazioni FOXC1, l’opacità corneale e la neovascolarizzazione sono più marcate, e il grado di anomalia corneale è maggiore rispetto alle mutazioni PITX2, con una maggiore frequenza di glaucoma1).
I reperti angolari includono inserzione alta dell’iride, residui uveali a corda e ispessimento della linea di Schwalbe (anello embrionale posteriore). Sono stati riportati anche casi con microsferofachia e sublussazione del cristallino7).
Il glaucoma complica il 50–60% dei casi. Un aumento della pressione intraoculare può verificarsi già nell’infanzia, ma nella maggior parte dei casi si manifesta nell’infanzia o in giovane età adulta. Alcuni casi vengono diagnosticati in occasione di un progressivo calo visivo; è importante non trascurare i reperti del segmento anteriore e le alterazioni glaucomatose6).
Li et al. (2021) hanno riportato un caso di un bambino di 7 anni (ARS tipo 3, mutazione de novo FOXC1) con diametro corneale di 14 mm, lunghezza assiale di 27,16/26,56 mm, rapporto C/D 0,9 e IOP 33/20 mmHg. È stato necessario un intervento chirurgico antiglaucoma bilaterale a partire dal 36° giorno di vita5).
I reperti sistemici sono i seguenti:
Circa il 50-60% dei pazienti con ARS sviluppa glaucoma. Si manifesta spesso durante l’infanzia o la giovane età adulta, ma può anche presentarsi nell’infanzia con aumento della pressione intraoculare. Sono necessarie misurazioni regolari della pressione intraoculare e valutazione del nervo ottico. Per maggiori dettagli, vedere la sezione «Trattamento standard».
L’ARS è una malattia autosomica dominante a penetranza completa. Tuttavia, anche all’interno della stessa famiglia, individui con la stessa mutazione genetica possono presentare quadri clinici molto diversi (espressività variabile)1).
In uno studio di coorte su larga scala, le mutazioni di FOXC1 e PITX2 sono state associate a un ampio spettro di glaucoma dall’infanzia all’età adulta9). Casi diagnosticati clinicamente come glaucoma congenito primario (PCG) possono essere riclassificati tramite test genetico. Quando i segni del segmento anteriore nei neonati sono sottili, il test genetico può contribuire a una diagnosi accurata del tipo di malattia.
Nei casi con microdelezione intorno al gene PITX2, la sovrapposizione delle delezioni di NEUROG2, UGT8 e NDST4 può causare ritardo dello sviluppo e disabilità intellettiva8)3).
Kawanami et al. (2023) hanno riportato il caso di un bambino giapponese di 3 anni con una microdelezione di 2,5 Mb in 4q25 (inclusi PITX2, NEUROG2 e ANK2). Presentava ernia ombelicale, coloboma dell’iride e ritardo dello sviluppo, ma l’ECG era normale nonostante la delezione di ANK2. L’aploinsufficienza di NEUROG2 è stata considerata una possibile causa del ritardo dello sviluppo8).
A causa dell’ereditarietà autosomica dominante, la probabilità di trasmissione da un genitore con mutazione è del 50%. La penetranza è completa, ma il fenotipo varia: la gravità dei sintomi può differire notevolmente per la stessa mutazione1). Si raccomandano test genetici e consulenza genetica.
La diagnosi di ARS si basa su anomalie bilaterali dell’angolo e dell’iride. La condizione diagnostica è la presenza di embriotossone posteriore con adesione parziale dell’iride periferica13). Se l’embriotossone posteriore non è confermabile alla lampada a fessura, è necessaria la gonioscopia. In caso di anomalie sistemiche, si richiede una valutazione pediatrica completa per la sindrome ARS13).
Si noti che un lieve embriotossone posteriore si osserva nell’8-15% della popolazione normale, ma da solo non è associato a glaucoma, ecc. L’anamnesi familiare è importante anche per la diagnosi.
Nella classificazione del glaucoma pediatrico secondo le Linee guida per il glaucoma (5a edizione), l’ARS è considerata un esempio tipico di glaucoma associato ad anomalie congenite dello sviluppo oculare13). La diagnosi viene posta quando anomalie dello sviluppo oculare presenti dalla nascita soddisfano i criteri diagnostici per il glaucoma pediatrico.
Le principali malattie da differenziare dall’ARS sono elencate di seguito.
| Malattia | Differenza dall’ARS |
|---|---|
| Sindrome ICE | Unilaterale, acquisita, predominanza femminile |
| Anomalia di Peters | Opacità corneale centrale, difetto della membrana di Descemet |
| Aniridia | Panno corneale, ipoplasia foveale |
| Distrofia corneale polimorfa posteriore | Bilaterale, familiare, nessuna differenza di sesso |
La sindrome ICE (atrofia progressiva dell’iride, sindrome di Chandler, ecc.) richiede una differenziazione dall’ARS, ma il punto di differenziazione principale è che l’ICE è unilaterale e acquisita, mentre l’ARS è bilaterale e congenita.
Attualmente non esiste una terapia curativa per l’ARS stessa. Il trattamento si concentra sulla gestione del glaucoma e sulla sorveglianza delle complicanze sistemiche. La strategia terapeutica segue quella del glaucoma congenito primario (PCG) 13).
Il glaucoma complica circa il 50-60% dei casi di ARS. La terapia farmacologica segue quella del glaucoma generale, ma spesso è inefficace.
Inibitori della produzione di umore acqueo
Beta-bloccanti : una delle opzioni di prima linea. Sicuri ed efficaci, ma spesso inefficaci nei bambini.
Inibitori dell’anidrasi carbonica (CAI) in collirio : brinzolamide, ecc. Possibile associazione con beta-bloccanti.
Alfa-2 agonisti (brimonidina) : controindicati al di sotto dei 2 anni per effetti neuropsichiatrici (apnea, bradicardia, ipotensione, ipotonia muscolare, depressione del SNC) 13).
Farmaci che favoriscono il deflusso dell'umore acqueo
Analoghi delle prostaglandine : latanoprost, travoprost, ecc. L’efficacia nei bambini è considerata inferiore rispetto agli adulti 13).
Esempio : In un bambino di 7 anni, controllo a lungo termine con travoprost + brinzolamide 5). In un uomo di 77 anni, la PIO era di 35 mmHg nonostante latanoprost, timololo e brinzolamide, indicando un controllo difficile 2).
Secondo un report, non vi è differenza di efficacia tra gli agonisti del recettore FP dei prostanoidi e i beta-bloccanti 13).
Nei neonati, la dose di collirio è relativamente alta rispetto al peso corporeo e alla superficie corporea, pertanto si dovrebbero utilizzare farmaci a concentrazione più bassa possibile 13).
Se il controllo della pressione intraoculare non viene ottenuto con la terapia farmacologica, si procede all’intervento chirurgico 10)13).
Le complicanze postoperatorie del GDD riportate includono: camera anteriore bassa 13,6%, ipotonia 11,7%, versamento coroideale 8,3%, endoftalmite 1,7% 14).
Chakraborty et al. (2022) hanno riportato un caso di distacco di retina associato a sindrome di Axenfeld-Rieger (ragazzo di 15 anni). Erano presenti microsferofachia e sublussazione del cristallino; dopo vitrectomia, la PIO è aumentata a 41 mmHg, formando uno stafiloma sclerale. È stata eseguita una ciclofotocoagulazione a diodo, ottenendo infine una PIO di 18 mmHg 7).
Il tasso di successo della chirurgia dell’angolo è inferiore a quello del PCG 13). Per la trabeculectomia con MMC, il tasso di successo a lungo termine a 2 anni è di circa il 59%; per i GDD, è riportato l’87% a 12 mesi e il 77% a 24 mesi 14). I casi refrattari possono richiedere più interventi chirurgici.
La causa fondamentale dell’ARS è un difetto nella migrazione e differenziazione delle cellule della cresta neurale. Lo sviluppo alterato delle cellule della cresta neurale nella camera anteriore, nell’angolo del segmento anteriore, nelle ossa facciali, nei denti, nel sistema cardiovascolare e nella pelle periombelicale porta a malformazioni multi-organo.
Alla fine del periodo fetale, le cellule endoteliali indifferenziate che rivestono la camera anteriore normalmente scompaiono dall’iride e dall’angolo. Nell’ARS, questo processo di scomparsa è alterato e le cellule endoteliali indifferenziate rimangono sull’iride, formando bande. Nell’angolo, si verifica un attacco alto dell’iride, che ricopre meccanicamente il trabecolato.
Istologicamente, si osserva un’estensione anomala di un monostrato di cellule endoteliali con una membrana simil-descemetica dalla superficie posteriore della cornea attraverso la camera anteriore, l’angolo e la superficie dell’iride. La membrana è presente nei quadranti con ectropion uveale e deviazione pupillare, mentre nei quadranti opposti si osserva atrofia dell’iride.
FOXC1 e PITX2 sono entrambi fattori di trascrizione che si legano a specifiche sequenze di DNA per regolare l’espressione dei geni a valle. Entrambi agiscono in sinergia nello sviluppo del segmento anteriore dell’occhio e regolano geni bersaglio comuni a valle 3). Il dominio forkhead di FOXC1 (un dominio legante il DNA di 110 amminoacidi) è il più importante dal punto di vista funzionale 2), e si suggerisce che le mutazioni in questo dominio siano più fortemente associate a sintomi neuropsichiatrici.
Vengono indicati due meccanismi di aumento della pressione intraoculare.
Il grado di assenza dell’iride e la quantità di processi iridei nell’angolo non sono necessariamente correlati alla gravità del glaucoma. Tuttavia, un alto grado di sinechie iridee nell’angolo predispone al glaucoma.
Le mutazioni FOXC1 promuovono il glaucoma congenito più di altre mutazioni 1), e le anomalie morfologiche del corpo ciliare e dell’angolo di drenaggio possono contribuire all’aumento della PIO 1).
Nei topi mutanti FOXC1 si osservano una diminuzione e anomalie strutturali delle fibre di collagene nello stroma corneale, nonché un danno alle cellule stromali corneali 1). Inoltre, FOXC1 agisce come inibitore della neovascolarizzazione corneale (attraverso il controllo della biodisponibilità del VEGF) 1); la perdita di questa inibizione a causa della mutazione FOXC1 porta a neovascolarizzazione corneale.
FOXC1, come fattore di trascrizione della famiglia FOX, svolge un ruolo importante anche nello sviluppo cerebrale 4).
Una revisione sistematica ha riportato che anomalie della sostanza bianca compaiono nel 41,3% dei casi di ARS 4). Le mutazioni FOXC1 possono indurre malattia dei piccoli vasi cerebrali (CSVD), iperintensità della sostanza bianca, spazi perivascolari dilatati, microemorragie e infarti lacunari.
Ohkubo et al. (2025) hanno confermato con RM cerebrale in un bambino giapponese di 2 anni (mutazione FOXC1: c.240del, p.Y81Ifs21) lesioni periventricolari della sostanza bianca, spazi perivascolari dilatati e tortuosità-dilatazione dell’arteria vertebrobasilare. Il padre aveva una storia di infarto cerebrale all’età di 18 anni 4).
Una revisione di 95 casi di mutazioni FOXC1 ha riscontrato sintomi neuropsichiatrici (difficoltà di apprendimento, epilessia, disabilità intellettiva, delirio di gelosia, ecc.) nel 6,3% dei casi, e l’83,3% dei casi con mutazioni nel dominio forkhead presentava sintomi neuropsichiatrici 2).
Sì. Una revisione sistematica indica che anomalie della sostanza bianca compaiono in circa il 41% dei casi di ARS con mutazione FOXC1 4). Le mutazioni FOXC1 potrebbero essere associate a un rischio di malattia dei piccoli vasi cerebrali e ictus; pertanto, un follow-up neurologico a lungo termine è importante, specialmente nell’ARS correlata a mutazioni FOXC1.
Negli ultimi anni, molte nuove mutazioni sono state riportate grazie al sequenziamento di nuova generazione e al sequenziamento dell’intero genoma.
Wowra et al. (2024) hanno identificato una grande delezione (nuova mutazione) che include una parte dell’esone 1 di FOXC1 e l’intera regione 3’UTR in tre sorelle polacche con ARS. I fenotipi erano molto diversi all’interno della stessa famiglia, e inizialmente era stata diagnosticata erroneamente la sindrome di Chandler 1).
Jiang et al. (2024) hanno identificato un riarrangiamento genomico complesso comprendente una delezione di 6,15 Mb del cromosoma 4q25 contenente PITX2, un’inversione di 45,71 Mb e una delezione di 14 bp in una famiglia cinese con ARS tipo 1. Una bambina di 11 anni presentava una PIO di 43,5/44,0 mmHg 3).
Altre segnalazioni di nuove mutazioni includono FOXC1 p.Phe136Leu (dominio forkhead) 2), FOXC1 p.S82R (mutazione de novo) 5) e FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino et al. (2024) hanno riportato il caso di un uomo giapponese di 77 anni con ARS tipo 3. A partire dai 72 anni sono comparsi deliri di gelosia ed è stata confermata una leucoencefalopatia. Una revisione della letteratura di 95 casi con mutazione FOXC1 ha mostrato che il 6,3% (6/95) presentava sintomi neuropsichiatrici, di cui l’83,3% (5/6) aveva una mutazione del dominio forkhead 2).
Questa osservazione suggerisce che il dominio funzionale della mutazione FOXC1 potrebbe essere coinvolto nella manifestazione di sintomi neuropsichiatrici, sottolineando l’importanza di un follow-up a lungo termine dal punto di vista della salute mentale.
È stato suggerito che le mutazioni FOXC1 possano indurre CSVD e aumentare il rischio di ictus 4). La prevenzione e l’intervento precoce delle malattie neurovascolari nei pazienti con ARS sono considerati argomenti di ricerca futuri.
La comprensione della patologia della «sclerizzazione» corneale dovuta a mutazioni FOXC1 sta progredendo 1). Si spera che la comprensione dei meccanismi molecolari dell’opacità corneale porti allo sviluppo di nuovi trattamenti che utilizzano terapia genica, farmaci antifibrotici e biomateriali 1).