ARS Tip 1
Sorumlu gen: PITX2 (4q25)
Başlıca anomaliler: Ön segment göz anomalileri, diş anomalileri, göbek çevresinde fazla deri/umbilikal herni, kraniyofasiyal anomaliler, kardiyovasküler anomaliler

Axenfeld-Rieger sendromu (ARS), ön segment disgenezisi ve sistemik anomalileri birleştiren konjenital bir hastalık grubudur. Temel neden, nöral krest hücrelerinin göç ve farklılaşma bozukluğudur. Fetal dönemin sonunda ön kamarayı kaplayan farklılaşmamış endotel hücrelerinin iristen ve açıdan normal olarak kaybolma süreci bozulur ve kalıntıları fibröz bantlar ve iris yapışıklıklarına yol açar.
Tarihsel arka plan: 1920’de Axenfeld, posterior embriyotokson (Schwalbe hattının öne yer değiştirmesi ve kalınlaşması) ve iris süreçlerini tanımladı. 1934-1935’te Rieger, iris hipoplazisi, pupil distopisi ve polikoriyi ek olarak bildirdi. Günümüzde üç aşamada sınıflandırılır:
Tüm bunlar Axenfeld-Rieger sendromu olarak adlandırılır. %50-60’ında glokom eşlik eder, otozomal dominant kalıtılır ve genellikle iki taraflıdır. Katarakt ve lens dislokasyonu da sık görülür.
Epidemiyoloji: Prevalans yaklaşık 1/200.000 olarak bildirilmiştir, ancak son raporlar 1/50.000 ila 100.000 arasında olduğunu tahmin etmektedir2)4). Cinsiyet farkı yoktur ve genellikle bebeklik veya erken çocukluk döneminde teşhis edilir.
Genetik sınıflandırma aşağıdaki gibidir:
ARS Tip 1
Sorumlu gen: PITX2 (4q25)
Başlıca anomaliler: Ön segment göz anomalileri, diş anomalileri, göbek çevresinde fazla deri/umbilikal herni, kraniyofasiyal anomaliler, kardiyovasküler anomaliler
ARS Tip 2
Sorumlu gen: 13q14 (doğrulanmamış)
Başlıca anomaliler: Ön segment göz anomalileri, glokom. Sistemik anomaliler Tip 1 ve Tip 3’e göre daha azdır.
ARS Tip 3
Sorumlu gen: FOXC1 (6p25)
Başlıca anomaliler: Ön segment göz anomalileri, glokom, sensörinöral işitme kaybı, atriyal septal defekt, böbrek anomalileri, beyaz cevher lezyonları
FOXC1 ve PITX2 mutasyonları ARS vakalarının %40-70’ini oluşturur5). Bununla birlikte, ARS’nin %60’ında sorumlu gen tanımlanamamıştır4) ve genetik çeşitlilik fazladır.
Çocukluk ve genç erişkin başlangıçlı glokomun büyük bir kayıt analizinde moleküler tanı oranı %56.5 bulunmuştur11). FOXC1 mutasyonu %20.3, PITX2 mutasyonu %17.4 ve PAX6 mutasyonu %10.1 oranında görülmüş olup, bilinen genlerle açıklanamayan vakalar az değildir11).
Sorumlu gene göre ayırt edilirler. Tip 1, PITX2 (4q25) mutasyonu ile diş, göbek ve yüz kemik anomalileri birlikteliğidir. Tip 3, FOXC1 (6p25) mutasyonu ile işitme kaybı, kalp defekti, böbrek ve nörolojik anomaliler birlikteliğidir. Tip 2, 13q14’te yer alır ancak sorumlu gen doğrulanmamıştır ve esas olarak ön segment anomalisi ve glokom ile karakterizedir. Genetik test ile kesin tanı konulabilir.
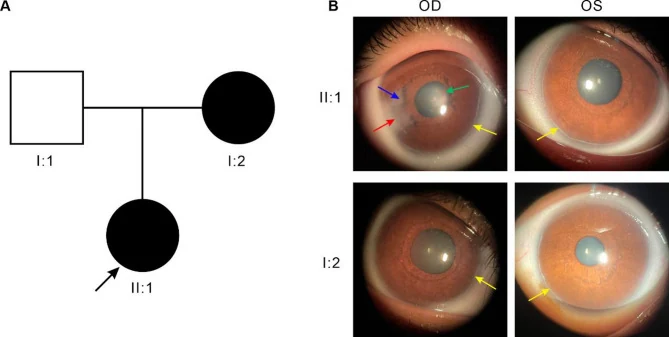
Göz bulgularının ana maddeleri aşağıda verilmiştir.
| Göz bulgusu | Özellik |
|---|---|
| Posterior embriyotokson | Schwalbe hattının öne yer değiştirmesi ve kalınlaşması |
| İris çıkıntıları | İpliksi ila geniş bant şeklinde |
| Pupil deviasyonu | Arka embriyotoksonun ters yönüne deviasyon |
| Yalancı polikori | İris stromasında delik benzeri görünüm |
| Uveal ektropion | İris pigment epitelinin dışa dönmesi |
Arka embriyotokson, Schwalbe hattında farklılaşmamış hücrelerin kalıntısıdır ve limbusdan 0.5-2.0 mm merkeze doğru, limbus boyunca çizgisel olarak görülür. Genellikle tüm çevre boyunca değil, bir kısımda sınırlıdır. Belirgin Schwalbe hattı ile iris yapışıklığı varsa Axenfeld anomalisi, buna iris stroma atrofisi eşlik ediyorsa Rieger anomalisi olarak adlandırılır.
Kornea genellikle saydamdır ve endotel yapısı normaldir, ancak kalıntı doku ile fiziksel temas sonucu sekonder kornea bulanıklığı oluşabilir. Kornea bulanıklığı genellikle periferle sınırlıdır ve genellikle görmeyi doğrudan etkilemez. Bununla birlikte, FOXC1 mutasyonunda kornea bulanıklığı ve kornea neovaskülarizasyonu daha belirgindir ve PITX2 mutasyonuna kıyasla kornea anormalliğinin derecesi daha büyük ve glokom sıklığı daha yüksektir 1).
Açı bulguları yüksek iris insersiyonu, kordiform uveal kalıntılar ve Schwalbe hattı kalınlaşması (arka embriyotokson) içerir. Mikrofaaki ve lens subluksasyonu birlikteliği de bildirilmiştir 7).
Glokom %50-60 oranında eşlik eder. Göz içi basıncı artışı bebeklik döneminde de ortaya çıkabilir, ancak çoğunlukla çocukluk- genç erişkinlik döneminde başlar. Progresif görme azalması ile tanı alan vakalar da vardır; ön segment bulguları ve glokomatöz değişiklikleri gözden kaçırmamak önemlidir 6).
Li ve ark. (2021) tarafından bildirilen 7 yaşında erkek çocuk (ARS tip 3, de novo FOXC1 mutasyonu) kornea çapı 14 mm, aksiyel uzunluk 27.16/26.56 mm, C/D oranı 0.9, GİB 33/20 mmHg idi. Doğumdan 36 gün sonra her iki göze antiglokom cerrahisi gerekti 5).
Sistemik bulgular aşağıdaki gibidir:
ARS’li bireylerin yaklaşık %50-60’ında glokom gelişir. Glokom genellikle çocukluktan genç erişkinliğe kadar ortaya çıkar, ancak bazı vakalarda bebeklik döneminde göz içi basıncı yüksekliği görülebilir. Düzenli göz içi basıncı ölçümü ve optik sinir değerlendirmesi gereklidir. Ayrıntılı bilgi için «Standart Tedavi Yöntemleri» bölümüne bakınız.
ARS otozomal dominant kalıtım gösterir ve penetransı tamdır. Ancak aynı aile içinde aynı gen mutasyonuna sahip bireylerde klinik bulgular büyük bireysel farklılıklar (değişken ifade) gösterebilir1).
Büyük kohort çalışmaları, FOXC1 ve PITX2 mutasyonlarının çocukluktan erişkinliğe kadar geniş bir glokom spektrumu ile ilişkili olduğunu göstermiştir9). Başlangıçta PCG (primer konjenital glokom) tanısı alan vakalar, genetik test sonrası yeniden sınıflandırılabilir. Bebeklerde ön segment bulguları hafif olduğunda, genetik test doğru hastalık tipi tanısına katkı sağlar.
PITX2 geni çevresinde mikrodelesyonu olan vakalarda, NEUROG2, UGT8 ve NDST4 delesyonlarının üst üste binmesi gelişim geriliği ve zihinsel engelliliğe neden olabilir8)3).
Kawanami ve ark. (2023), 4q25’te 2.5 Mb’lik bir mikrodelesyon (PITX2, NEUROG2 ve ANK2 dahil) taşıyan 3 yaşında bir Japon erkek çocuğu bildirdi. Çocukta omfalosel, iris kolobomu ve gelişim geriliği vardı, ancak ANK2 delesyonuna rağmen EKG normaldi. NEUROG2’nin haployetersizliği gelişim geriliğinin olası nedeni olarak değerlendirildi8).
Otozomal dominant kalıtım nedeniyle, mutasyon taşıyan ebeveynden geçme olasılığı %50’dir. Penetrans tamdır ancak fenotip değişkendir; aynı mutasyonla semptomların şiddeti büyük ölçüde farklılık gösterebilir1). Genetik test ve genetik danışmanlık önerilir.
ARS tanısı bilateral açı ve iris anomalilerine dayanır. Posterior embriyotoksona periferik irisin kısmi yapışması tanı kriteri olarak kabul edilir13). Yarık lamba ile posterior embriyotoksonun görülemediği durumlarda gonyoskopi gereklidir. Sistemik anormallikler varsa, ARS sendromu olarak pediatriye tam sistemik inceleme için sevk edilir13).
Normal popülasyonun %8-15’inde hafif posterior embriyotokson görülebilir, ancak tek başına glokom vb. eşlik etmez. Aile öyküsü almak da tanıda önemlidir.
Glokom Klinik Kılavuzu (5. baskı) pediatrik glokom sınıflandırmasında ARS, konjenital oküler gelişim anomalileri ile ilişkili glokomun tipik bir örneği olarak yer alır13). Doğumdan itibaren var olan oküler anomali, pediatrik glokom tanı kriterlerini karşılıyorsa tanı konur.
ARS’den ayırt edilmesi gereken başlıca hastalıklar aşağıda gösterilmiştir.
| Hastalık | ARS’den farkı |
|---|---|
| ICE sendromu | Tek taraflı, edinsel, kadın baskın |
| Peters anomalisi | Santral kornea opasitesi, Descemet membran defekti |
| Aniridi | Korneal pannus, foveal hipoplazi |
| Posterior polimorföz korneal distrofi | Bilateral, ailesel, cinsiyet farkı yok |
ICE sendromu (progresif iris atrofisi, Chandler sendromu vb.) ARS’den ayırt edilmelidir; ancak ICE tek taraflı ve edinsel iken ARS bilateral ve konjenitaldir, bu en önemli ayırıcı noktadır.
ARS’nin kendisi için şu anda küratif bir tedavi yoktur; tedavinin temelini glokom yönetimi ve sistemik komplikasyonların takibi oluşturur. Tedavi stratejisi erken başlangıçlı gelişimsel glokoma (primer konjenital glokom: PCG) benzer13).
Glokom, ARS vakalarının yaklaşık %50-60’ına eşlik eder. İlaç tedavisi genel glokoma benzer şekilde uygulanır, ancak çoğu zaman etkisizdir.
Aköz hümör üretimini baskılayan ilaçlar
Beta blokerler: Birinci basamak seçeneklerden biridir. Güvenli ve etkilidir, ancak çocuklarda sıklıkla etkisizdir.
Karbonik anhidraz inhibitörleri (CAI) damla: Brinzolamid gibi. Beta blokerlerle birlikte kullanılabilir.
Alfa-2 agonistleri (brimonidin): 2 yaş altında nöropsikiyatrik yan etkiler (apne, bradikardi, hipotansiyon, kas hipotonisi, santral sinir sistemi depresyonu) nedeniyle kontrendikedir13).
Aköz hümör çıkışını artıran ilaçlar
Prostaglandin analogları: Latanoprost, travoprost gibi. Çocuklarda etkinlik yetişkinlere göre daha zayıftır13).
Örnek vaka: 7 yaşında erkek çocukta travoprost + brinzolamid ile uzun süreli yönetim sağlanmıştır5). 77 yaşında erkek hastada latanoprost/timolol + brinzolamid ile GİB 35 mmHg’de kalmış ve kontrolü zor olmuştur2).
Prostanoid FP reseptör agonistleri ile beta blokerler arasında etkinlik farkı olmadığı bildirilmiştir13).
Bebeklerde ve küçük çocuklarda vücut ağırlığı ve yüzey alanına göre damla dozu nispeten yüksek olduğundan, mümkün olduğunca düşük konsantrasyonlu ilaçlar kullanılmalıdır13).
İlaç tedavisi ile göz içi basıncı kontrol altına alınamazsa cerrahi uygulanır10)13).
GDD cerrahisi sonrası komplikasyonlar: ön kamara sığlaşması %13,6, hipotoni %11,7, koroid efüzyonu %8,3, endoftalmi %1,7 olarak bildirilmiştir14).
Chakraborty ve ark. (2022), ARS’ye eşlik eden retina dekolmanı (15 yaşında erkek) olgusunu bildirmiştir. Mikrosferofaki ve lens subluksasyonu mevcuttu, vitrektomi sonrası GİB 41 mmHg’ye yükseldi ve stafilom oluştu. Diyot silier fotokoagülasyon uygulandı ve sonunda GİB 18 mmHg’ye ulaşıldı7).
Açı cerrahisinin başarı oranı PCG’den daha düşüktür 13). MMC ile kombine trabekülektomide 2 yıllık uzun dönem başarı oranı yaklaşık %59, GDD’de ise 12. ayda %87 ve 24. ayda %77 olarak bildirilmiştir 14). Dirençli olgularda birden fazla cerrahi gerekebilir.
ARS’nin temel nedeni, nöral krest hücrelerinin göç ve farklılaşma kusurudur. Ön kamara, ön segment açısı, yüz kemikleri, dişler, kardiyovasküler sistem ve göbek çevresi ciltte nöral krest hücre gelişiminin bozulması çoklu organ malformasyonlarına yol açar.
Fetal dönemin sonunda, normalde ön kamarayı kaplayan farklılaşmamış endotel hücreleri iristen ve açıdan kaybolur. ARS’de bu kaybolma süreci bozulur ve farklılaşmamış endotel hücreleri iris üzerinde kalarak bant oluşumuna neden olur. Açıda iris yüksek yapışıklığı oluşur ve trabekülümü mekanik olarak örter.
Histolojik olarak, kornea arka yüzeyinden ön kamara, açı ve iris yüzeyine kadar Descemet benzeri bir membranla birlikte tek katlı endotel benzeri hücre tabakası anormal şekilde yayılır. Bu membran, üveal ektropiyon ve pupil deplasmanı olan kadranlarda bulunurken, karşı kadranda iris atrofisi görülür.
FOXC1 ve PITX2, belirli DNA dizilerine bağlanarak aşağı akış genlerinin ekspresyonunu düzenleyen transkripsiyon faktörleridir. Her ikisi de ön segment gelişiminde sinerjistik olarak etki eder ve ortak aşağı akış hedef genlerini düzenler 3). FOXC1’in forkhead alanı (110 amino asitlik DNA bağlanma alanı) işlevsel olarak en önemlisidir 2) ve bu alandaki mutasyonların nöropsikiyatrik semptomlarla daha güçlü ilişkili olduğu düşünülmektedir.
Göz içi basınç artışının mekanizması olarak aşağıdaki iki durum belirtilmektedir:
İris defektinin derecesi ve açıdaki iris çıkıntılarının miktarı, glokom şiddeti ile her zaman korele değildir. Ancak açıdaki iris yapışıklığının derecesi yüksekse glokoma yatkınlık artar.
FOXC1 mutasyonu, diğer mutasyonlara kıyasla konjenital glokomu daha fazla tetikler 1) ve siliyer cisim ile drenaj açısındaki morfolojik anormallikler GİB artışına katkıda bulunabilir 1).
FOXC1 mutant farelerde, kornea stromasında kollajen liflerinde azalma ve yapısal anormallik ile stromal hücre hasarı gözlenir 1). Ayrıca FOXC1, kornea neovaskülarizasyonunun baskılayıcısı olarak işlev görür (VEGF’nin biyoyararlanımını kontrol ederek) 1) ve FOXC1 mutasyonu ile bu baskılamanın kaybı kornea neovaskülarizasyonuna yol açar.
FOXC1, FOX ailesi transkripsiyon faktörü olarak beyin gelişiminde de önemli bir rol oynar 4).
Sistematik bir derlemede, ARS’li vakaların %41,3’ünde beyaz cevher anormallikleri rapor edilmiştir 4). FOXC1 mutasyonu, serebral küçük damar hastalığı, beyaz cevher hiperintensiteleri, genişlemiş perivasküler boşluklar, mikrokanamalar ve laküner enfarktüse neden olabilir.
Ohkubo ve ark. (2025), 2 yaşında bir Japon erkek çocuğunda (FOXC1 mutasyonu: c.240del, p.Y81Ifs21) beyin MRG’sinde periventriküler beyaz cevher lezyonları, genişlemiş perivasküler boşluklar ve vertebrobaziler arterlerde kıvrımlı genişleme doğruladı. Babasında 18 yaşında serebral enfarktüs öyküsü vardı 4).
95 FOXC1 mutasyonu vakasının derlemesinde %6,3’ünde nöropsikiyatrik semptomlar (öğrenme güçlüğü, epilepsi, zihinsel engel, kıskançlık sanrıları vb.) görülmüş ve forkhead alan mutasyonu olan vakaların %83,3’ünde nöropsikiyatrik semptomlar ortaya çıkmıştır 2).
Evet. FOXC1 mutasyonlu ARS’de beyaz cevher anormalliklerinin yaklaşık %41 vakada görüldüğüne dair sistematik bir derleme vardır 4). FOXC1 mutasyonunun serebral küçük damar hastalığı ve inme riski ile ilişkili olabileceği belirtilmektedir; özellikle FOXC1 mutant ARS’de nörolojik uzun dönem takip önemlidir.
Son yıllarda, yeni nesil dizileme ve tüm genom dizileme ile birçok yeni mutasyon rapor edilmiştir.
Wowra ve ark. (2024), ARS’li üç Polonyalı kız kardeşte FOXC1 ekzon 1’in bir kısmını ve 3’UTR’nin tamamını içeren büyük bir delesyon (yeni mutasyon) tanımladı. Aynı ailede fenotipler önemli ölçüde farklıydı ve başlangıçta yanlışlıkla Chandler sendromu tanısı konmuştu1).
Jiang ve ark. (2024), ARS tip 1’li bir Çinli ailede PITX2’yi içeren 6.15 Mb’lik kromozom 4q25 delesyonu, 45.71 Mb’lik inversiyon ve 14 bp’lik delesyondan oluşan karmaşık bir genomik yeniden düzenleme tanımladı. 11 yaşındaki bir kız çocuğunda GİB 43.5/44.0 mmHg idi3).
Diğer yeni mutasyon raporları arasında FOXC1 p.Phe136Leu (forkhead alanı)2), FOXC1 p.S82R (de novo mutasyon)5) ve FOXC1 c.240del, p.Y81Ifs214) yer almaktadır.
Yoshino ve ark. (2024), 77 yaşında bir Japon erkekte ARS tip 3 vakası bildirdi. 72 yaşından itibaren kıskançlık sanrıları ortaya çıktı ve lökensefalopati doğrulandı. FOXC1 mutasyonlu 95 vakanın literatür taramasında %6,3’ünde (6/95) nöropsikiyatrik semptomlar görüldü ve bunların %83,3’ünde (5/6) forkhead alan mutasyonu vardı2).
Bu bulgu, FOXC1 mutasyonunun fonksiyonel alanının nöropsikiyatrik semptomların ortaya çıkmasında rol oynayabileceğini göstermekte ve ruh sağlığı açısından uzun süreli takibin önemini vurgulamaktadır.
FOXC1 mutasyonunun CSVD’yi indükleyerek inme riskini artırabileceği belirtilmiştir4). ARS hastalarında nörovasküler hastalıkların önlenmesi ve erken müdahalesinin önemi gelecekteki araştırma konuları arasındadır.
FOXC1 mutasyonuna bağlı korneanın “skleralizasyonu”nun patogenezi anlaşılmaktadır1). Kornea opasitesinin moleküler mekanizmasının anlaşılmasının, gen tedavisi, anti-fibrotik ilaçlar ve biyomalzemeler kullanılarak yeni tedavilerin geliştirilmesine yol açması beklenmektedir1).