ARS النوع 1
الجين المسبب: PITX2 (4q25)
الاضطرابات الرئيسية: تشوهات الجزء الأمامي للعين، تشوهات الأسنان، الجلد الزائد حول السرة/الفتق السري، تشوهات القحف الوجهي، تشوهات القلب والأوعية الدموية

متلازمة أكسنفيلد-ريغر (ARS) هي مجموعة من الأمراض الخلقية التي تجمع بين تشوهات الجزء الأمامي من العين وتشوهات جهازية. السبب الأساسي هو خلل في هجرة وتمايز خلايا العرف العصبي. في نهاية فترة الحمل، تتعطل عملية اختفاء الخلايا البطانية غير المتمايزة التي تغطي الحجرة الأمامية من القزحية والزاوية، مما يؤدي إلى بقائها وتشكيل حبال أو التصاق مرتفع للقزحية.
خلفية تاريخية: في عام 1920، وصف أكسنفيلد حلقة شفالبه الخلفية (إزاحة أمامية وتثخن لخط شفالبه) ونتوءات القزحية. في 1934-1935، أضاف ريغر نقص تنسج القزحية، انحراف حدقة العين، وتعدد الحدقات. يُصنف الآن إلى ثلاث مراحل:
يُشار إلى هذه مجتمعة باسم متلازمة أكسنفيلد-ريغر. 50-60% منها يصاحبها زرق، وهي وراثية جسمية سائدة وعادة ما تكون ثنائية الجانب. كما تترافق بشكل كبير مع إعتام عدسة العين وخلع العدسة.
علم الأوبئة: كان معدل الانتشار يُقدر بحوالي 1/200,000، لكن التقارير الحديثة تشير إلى تقديرات تتراوح بين 1/50,000 و1/100,0002)4). لا يوجد تفاوت بين الجنسين، ويتم تشخيصه غالبًا في مرحلة الرضاعة والطفولة المبكرة.
التصنيف الوراثي هو كما يلي:
ARS النوع 1
الجين المسبب: PITX2 (4q25)
الاضطرابات الرئيسية: تشوهات الجزء الأمامي للعين، تشوهات الأسنان، الجلد الزائد حول السرة/الفتق السري، تشوهات القحف الوجهي، تشوهات القلب والأوعية الدموية
ARS النوع 2
الجين المسبب: 13q14 (غير مؤكد)
الاضطرابات الرئيسية: تشوهات الجزء الأمامي للعين، الجلوكوما. الاضطرابات الجهازية أقل من النوعين 1 و3
ARS النوع 3
الجين المسبب: FOXC1 (6p25)
الاضطرابات الرئيسية: تشوهات الجزء الأمامي للعين، الجلوكوما، فقدان السمع الحسي العصبي، عيب الحاجز الأذيني، تشوهات الكلى، آفات المادة البيضاء
تشكل طفرات FOXC1 وPITX2 40-70% من حالات ARS5). ومع ذلك، في 60% من حالات ARS، لا يزال الجين المسبب غير معروف4)، مما يشير إلى تنوع وراثي كبير.
في تحليل سجل كبير للجلوكوما لدى الأطفال والشباب، بلغت نسبة التشخيص الجزيئي 56.5%11). شكلت طفرات FOXC1 20.3%، وPITX2 17.4%، وPAX6 10.1%، ولا تزال هناك حالات لا يمكن تفسيرها بالجينات المعروفة11).
يتم التمييز بناءً على الجين المسبب. النوع 1 ناتج عن طفرة في PITX2 (4q25) ويصاحبه تشوهات في الأسنان والسرة وعظام الوجه. النوع 3 ناتج عن طفرة في FOXC1 (6p25) ويصاحبه فقدان السمع وعيوب القلب وتشوهات الكلى والاضطرابات العصبية. النوع 2 يقع في 13q14 لكن الجين المسبب غير مؤكد، وتكون تشوهات الجزء الأمامي للعين والجلوكوما هي السمات الرئيسية. يمكن تأكيد التشخيص بالاختبار الجيني.
العناصر الرئيسية لنتائج العين موضحة أدناه.
| نتيجة العين | الخصائص |
|---|---|
| الحلقة الجنينية الخلفية | انزياح خط شفالبي للأمام وسماكته |
| نتوءات القزحية | رفيعة خيطية إلى عريضة شريطية |
| انحراف حدقة | انحراف في الاتجاه المعاكس للحلقة الجنينية الخلفية |
| تعدد الحدقات الكاذب | مظهر ثقبي في سدى القزحية |
| انقلاب عنبي | انقلاب ظهارة القزحية الصبغية |
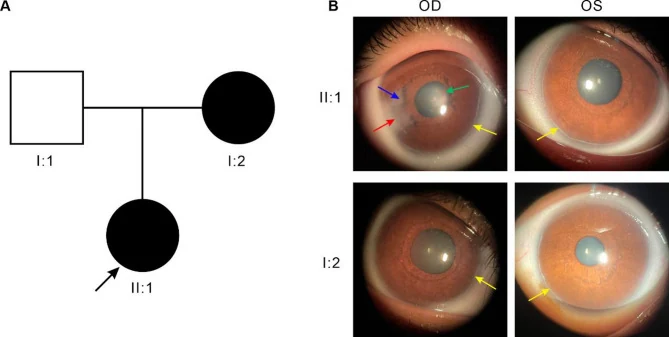
الحلقة الجنينية الخلفية هي بقايا خلايا غير متمايزة في خط شفالبه، وتظهر خطيًا على طول الحوف على بعد 0.5-2.0 مم إلى الداخل من الحوف. غالبًا ما تكون موضعية جزئيًا وليست محيطية بالكامل. إذا التصق خط شفالبه البارز بالقزحية، يُسمى شذوذ أكسنفيلد، وإذا صاحبه ضمور في سدى القزحية، يُسمى شذوذ ريجر.
القرنية عادة ما تكون شفافة وبنية البطانة طبيعية، ولكن قد يحدث عتامة قرنية ثانوية بسبب التلامس الفيزيائي مع الأنسجة المتبقية. غالبًا ما تكون العتامة موضعية في المنطقة المحيطية ولا تؤثر مباشرة على الرؤية. ومع ذلك، في طفرات FOXC1، تكون عتامة القرنية وتوعيتها أكثر وضوحًا، ويكون مدى شذوذ القرنية أكبر وتواتر الجلوكوما أعلى مقارنة بطفرات PITX21).
في فحص الزاوية، نجد ارتباطًا مرتفعًا للقزحية، وبقايا عنبية حبلية، وتثخن خط شفالبه (الحلقة الجنينية الخلفية). كما تم الإبلاغ عن حالات مصحوبة بعدسة صغيرة كروية أو خلع جزئي للعدسة7).
الجلوكوما تحدث في 50-60% من الحالات. قد يحدث ارتفاع ضغط العين منذ الطفولة المبكرة، لكن معظم الحالات تظهر في مرحلة الطفولة أو الشباب. قد يتم تشخيصها عند تدهور الرؤية التدريجي، لذا من المهم عدم إغفال علامات الجلوكوما ونتائج فحص الجزء الأمامي6).
في حالة Li et al. (2021) لصبي عمره 7 سنوات (ARS نوع 3، طفرة de novo FOXC1)، كان قطر القرنية 14 مم، طول المحور 27.16/26.56 مم، نسبة C/D 0.9، ضغط العين 33/20 مم زئبق. تطلبت كلتا العينين جراحة مضادة للجلوكوما منذ اليوم 36 بعد الولادة5).
النتائج الجهازية هي كما يلي:
حوالي 50-60% من مرضى متلازمة أكسينفيلد-ريجر يصابون بالجلوكوما. غالبًا ما يبدأ في مرحلة الطفولة إلى سن الشباب، ولكن هناك حالات يحدث فيها ارتفاع ضغط العين منذ الطفولة المبكرة. يلزم قياس ضغط العين بانتظام وتقييم العصب البصري. لمزيد من التفاصيل، راجع قسم “طرق العلاج القياسية”.
متلازمة أكسينفيلد-ريجر هي وراثة جسمية سائدة مع نفاذية كاملة. ومع ذلك، حتى داخل نفس العائلة التي تحمل نفس الطفرة الجينية، يمكن أن يكون هناك تباين كبير في المظاهر السريرية (تعبيرية متغيرة)1).
في دراسة أترابية كبيرة، ارتبطت طفرات FOXC1 وPITX2 بطيف واسع من الجلوكوما من الطفولة إلى البلوغ9). قد يتم إعادة تصنيف الحالات التي تم تشخيصها سريريًا في البداية على أنها جلوكوما خلقية أولية (PCG) عن طريق الاختبار الجيني، ويساهم الاختبار الجيني في التشخيص الدقيق للنوع عندما تكون نتائج الجزء الأمامي للعين لدى الرضع غير واضحة.
في الحالات التي يوجد فيها حذف صغير حول جين PITX2، قد يؤدي تداخل حذف NEUROG2 وUGT8 وNDST4 إلى تأخر النمو والإعاقة الذهنية 8)3).
أبلغ Kawanami et al. (2023) عن طفل ياباني يبلغ من العمر 3 سنوات لديه حذف صغير بحجم 2.5 ميغاباز في 4q25 (يشمل PITX2 وNEUROG2 وANK2). ظهرت لديه فتق سري، وورم قزحي، وتأخر في النمو، ولكن كان مخطط كهربية القلب طبيعيًا على الرغم من حذف ANK2. تم اعتبار نقص جرعة NEUROG2 كسبب محتمل لتأخر النمو 8).
نظرًا لأنها صفة وراثية سائدة، فإن احتمال وراثة الطفرة من أحد الوالدين هو 50%. الاختراقية كاملة ولكن النمط الظاهري يختلف بين الأفراد، وقد تختلف شدة الأعراض بشكل كبير حتى مع نفس الطفرة 1). يُوصى بإجراء الفحص الجيني والاستشارة الوراثية.
يعتمد تشخيص ARS على وجود شذوذ في زاوية العين والقزحية في كلتا العينين. يُعتبر وجود الحلقة الجنينية الخلفية (posterior embryotoxon) مع التصاق جزئي للقزحية المحيطة بها شرطًا للتشخيص 13). إذا لم يتم رؤية الحلقة الجنينية الخلفية بواسطة المصباح الشقي، يلزم إجراء فحص بزاوية العين (gonioscopy). في حالة وجود تشوهات جهازية، يتم تحويل المريض إلى طبيب أطفال لإجراء فحص شامل كجزء من متلازمة ARS 13).
تجدر الإشارة إلى أن 8-15% من الأشخاص الطبيعيين لديهم حلقة جنينية خلفية طفيفة، ولكنها وحدها لا تترافق مع الجلوكوما أو غيره. كما أن أخذ التاريخ العائلي مهم في التشخيص.
في نظام تصنيف الجلوكوما لدى الأطفال وفقًا لإرشادات علاج الجلوكوما (الطبعة الخامسة)، تُصنف ARS كمثال نموذجي لـ الجلوكوما المرتبطة بتشوهات العين الخلقية 13). يتم التشخيص عندما تستوفي تشوهات العين الموجودة منذ الولادة معايير تشخيص الجلوكوما لدى الأطفال.
فيما يلي الأمراض الرئيسية التي يجب تمييزها عن متلازمة أكسفيلد-ريجر (ARS).
| المرض | الاختلاف عن ARS |
|---|---|
| متلازمة القزحية القرنية البطانية (ICE) | أحادي الجانب، مكتسب، غلبة عند الإناث |
| شذوذ بيترز | عتامة مركز القرنية، نقص غشاء ديسيميه |
| انعدام القزحية | توعية القرنية، نقص تنسج النقرة |
| حثل القرنية متعدد الأشكال الخلفي | ثنائي الجانب، عائلي، لا فرق بين الجنسين |
من المهم التمييز بين متلازمة ICE (مثل ضمور القزحية التدريجي، متلازمة تشاندلر) وARS، ولكن الفرق الرئيسي هو أن ICE أحادية الجانب ومكتسبة بينما ARS ثنائية الجانب وخلقية.
لا يوجد حاليًا علاج جذري لـ ARS نفسه، ويركز العلاج على إدارة الجلوكوما ومراقبة المضاعفات الجهازية. يتبع نهج العلاج الجلوكوما التطورية المبكرة (الجلوكوما الخلقية الأولية: PCG) 13).
تحدث الجلوكوما في حوالي 50-60% من حالات ARS. يتم العلاج الدوائي وفقًا لإرشادات الجلوكوما العامة، لكنه غالبًا ما يكون غير فعال.
مثبطات إنتاج الخلط المائي
حاصرات بيتا: أحد الخيارات الأولى. آمنة وفعالة ولكنها غالبًا غير فعالة عند الأطفال.
قطرات مثبطات الأنهيدراز الكربونيك (CAI): مثل برينزولاميد. يمكن استخدامها مع حاصرات بيتا.
ناهضات ألفا-2 (بريمونيدين): ممنوعة للأطفال دون سن الثانية بسبب الآثار العصبية النفسية (انقطاع النفس، بطء القلب، انخفاض ضغط الدم، نقص التوتر العضلي، تثبيط الجهاز العصبي المركزي) 13).
أدوية تعزيز تدفق الخلط المائي
نظائر البروستاجلاندين: مثل لاتانوبروست وترافوبروست. تأثيرها عند الأطفال أضعف مقارنة بالبالغين 13).
مثال: في حالة صبي يبلغ من العمر 7 سنوات، تمت الإدارة طويلة المدى باستخدام ترافوبروست + برينزولاميد 5). في حالة رجل يبلغ من العمر 77 عامًا، كان ضغط العين 35 مم زئبق حتى مع لاتانوبروست/تيمولول + برينزولاميد، مما يشير إلى صعوبة السيطرة 2).
هناك تقارير تفيد بعدم وجود فرق في الفعالية بين ناهضات مستقبلات FP البروستانويد وحاصرات بيتا 13).
عند الرضع والأطفال الصغار، تكون جرعة القطرات كبيرة نسبيًا مقارنة بالوزن ومساحة سطح الجسم، لذا يجب استخدام أقل تركيز ممكن من الدواء 13).
إذا لم يتم التحكم في ضغط العين بالأدوية، يتم إجراء الجراحة10)13).
تشمل مضاعفات ما بعد زراعة GDD: ضحالة الغرفة الأمامية بنسبة 13.6%، انخفاض ضغط العين بنسبة 11.7%، انصباب مشيمي بنسبة 8.3%، والتهاب باطن العين بنسبة 1.7%14).
أبلغ Chakraborty وآخرون (2022) عن حالة انفصال الشبكية المرتبط بمتلازمة أكسفيلد-ريجر (طفل يبلغ من العمر 15 عامًا). كان مصحوبًا بعدسة كروية صغيرة وخلع جزئي للعدسة، وبعد استئصال الزجاجية ارتفع ضغط العين إلى 41 مم زئبق وتشكل قيلة صلبة. تم إجراء تخثير الجسم الهدبي بالديود وتم الوصول في النهاية إلى ضغط عين 18 مم زئبق7).
معدل نجاح جراحة الزاوية أقل من الجلوكوما الخلقي الأولي 13). في استئصال التربيق مع الميتومايسين سي، يبلغ معدل النجاح طويل الأمد لمدة عامين حوالي 59%، وفي أجهزة تصريف الجلوكوما، يبلغ 87% عند 12 شهرًا و77% عند 24 شهرًا 14). قد تتطلب الحالات المقاومة عمليات جراحية متعددة.
السبب الأساسي لمتلازمة أكسفيلد-ريجر هو خلل في هجرة وتمايز خلايا العرف العصبي. يؤدي ضعف نمو خلايا العرف العصبي في الغرفة الأمامية، زاوية العين الأمامية، عظام الوجه، الأسنان، الجهاز القلبي الوعائي، والجلد حول السرة إلى تشوهات متعددة الأعضاء.
في نهاية فترة الحمل، تختفي الخلايا البطانية غير المتمايزة التي تغطي الغرفة الأمامية بشكل طبيعي من القزحية والزاوية. في متلازمة أكسفيلد-ريجر، تتعطل عملية الاختفاء هذه، وتبقى الخلايا البطانية غير المتمايزة على القزحية مسببة تكوين حبال. في الزاوية، يحدث التصاق مرتفع للقزحية، مما يغطي التربيق ميكانيكيًا.
نسيجيًا، توجد طبقة أحادية من الخلايا البطانية الشبيهة بالبطانة مع غشاء يشبه غشاء ديسيميه تمتد بشكل غير طبيعي من السطح الخلفي للقرنية عبر الغرفة الأمامية والزاوية وسطح القزحية. يوجد الغشاء في الأرباع التي تعاني من انقلاب العنبية وانحراف الحدقة، بينما يُلاحظ ضمور القزحية في الأرباع المقابلة.
كل من FOXC1 وPITX2 هما عاملان نسخيان، يرتبطان بتسلسلات DNA محددة وينظمان التعبير الجيني النهائي. يعملان بشكل تآزري في تطور الجزء الأمامي للعين وينظمان جينات مستهدفة مشتركة 3). مجال الشوكة لـ FOXC1 (مجال ربط DNA مكون من 110 حمض أميني) هو الأكثر أهمية وظيفيًا 2)، وقد تشير طفرات هذا المجال إلى ارتباط أقوى بالأعراض العصبية النفسية.
تم تحديد آليتين لارتفاع ضغط العين:
لا يرتبط مدى نقص القزحية أو كمية نتوءات القزحية الزاوية بالضرورة مع شدة الجلوكوما. ومع ذلك، فإن الالتصاق القزحي الشديد في الزاوية يزيد من خطر الإصابة بالجلوكوما.
تحفز طفرة FOXC1 الجلوكوما الخلقية أكثر من الطفرات الأخرى 1)، وقد تساهم التشوهات الشكلية للجسم الهدبي وزاوية التصريف في ارتفاع ضغط العين 1).
في الفئران التي تحمل طفرة FOXC1، لوحظ انخفاض في ألياف الكولاجين في سدى القرنية وتشوهات بنيوية وتلف في خلايا السدى 1). بالإضافة إلى ذلك، يعمل FOXC1 كعامل مثبط لتكوين الأوعية الدموية في القرنية (من خلال التحكم في التوافر الحيوي لـ VEGF) 1)، ويؤدي فقدان هذا التثبيط بسبب طفرة FOXC1 إلى تكوين أوعية دموية في القرنية.
يلعب FOXC1، باعتباره عامل نسخ من عائلة FOX، دورًا مهمًا أيضًا في تطور الدماغ 4).
في مراجعة منهجية، تم الإبلاغ عن ظهور تشوهات المادة البيضاء في 41.3% من حالات متلازمة أكسفيلد-ريجر 4). يمكن أن تحفز طفرة FOXC1 مرض الأوعية الدموية الدماغية الصغيرة، وفرط كثافة المادة البيضاء، وتوسع المساحات المحيطة بالأوعية، والنزيف الدقيق، والاحتشاء الجوبي.
أكد Ohkubo وآخرون (2025) وجود آفات المادة البيضاء حول البطين، وتوسع المساحات المحيطة بالأوعية، والتعرج والتوسع في الشريان الفقري القاعدي في التصوير بالرنين المغناطيسي للدماغ لطفل ياباني يبلغ من العمر عامين (طفرة FOXC1: c.240del, p.Y81Ifs21). كان لدى والده تاريخ من احتشاء دماغي في سن 18 عامًا 4).
في مراجعة لـ 95 حالة من طفرات FOXC1، تم العثور على أعراض نفسية عصبية (صعوبات التعلم، الصرع، الإعاقة الذهنية، الغيرة الوهمية، إلخ) في 6.3%، وظهرت الأعراض النفسية العصبية في 83.3% من الحالات التي تحمل طفرات في مجال الشوكة الرأسية 2).
نعم. هناك مراجعة منهجية تشير إلى أن تشوهات المادة البيضاء تظهر في حوالي 41% من حالات متلازمة أكسفيلد-ريجر التي تحمل طفرة FOXC1 4). يُشار إلى أن طفرة FOXC1 قد ترتبط بمرض الأوعية الدموية الدماغية الصغيرة وخطر السكتة الدماغية، وخاصة في متلازمة أكسفيلد-ريجر المرتبطة بـ FOXC1، فإن المتابعة العصبية طويلة المدى مهمة.
في السنوات الأخيرة، تم الإبلاغ عن العديد من الطفرات الجديدة من خلال تسلسل الجيل التالي وتسلسل الجينوم الكامل.
حدد Wowra وآخرون (2024) حذفًا كبيرًا (طفرة جديدة) يشمل جزءًا من إكسون 1 ومنطقة 3’UTR بأكملها من جين FOXC1 في ثلاث شقيقات بولنديات مصابات بمتلازمة أكسفيلد-ريجر. اختلف النمط الظاهري بشكل كبير حتى داخل نفس العائلة، وتم تشخيصهن خطأً في البداية بمتلازمة تشاندلر 1).
حدد Jiang وآخرون (2024) إعادة ترتيب جينومي معقد يتكون من حذف 6.15 ميغاباز في الكروموسوم 4q25 يشمل PITX2، وانقلاب 45.71 ميغاباز، وحذف 14 زوجًا قاعديًا في عائلة صينية مصابة بمتلازمة أكسفيلد-ريجر من النوع 1. أظهرت فتاة تبلغ من العمر 11 عامًا ضغط عين يبلغ 43.5/44.0 مم زئبق 3).
تشمل التقارير الأخرى عن الطفرات الجديدة FOXC1 p.Phe136Leu (مجال رأس الشوكة) 2)، وFOXC1 p.S82R (طفرة جديدة) 5)، وFOXC1 c.240del, p.Y81Ifs21 4).
أبلغ Yoshino وآخرون (2024) عن حالة لرجل ياباني يبلغ من العمر 77 عامًا مصاب بمتلازمة أكسفيلد-ريجر من النوع 3. ظهرت أوهام الغيرة منذ سن 72 عامًا، وتم تأكيد اعتلال بيضاء الدماغ. في مراجعة أدبية لـ 95 حالة من طفرات FOXC1، لوحظت أعراض نفسية عصبية في 6.3% (6/95)، وكان 83.3% (5/6) منهم لديهم طفرات في مجال رأس الشوكة 2).
تشير هذه النتائج إلى أن المجال الوظيفي لطفرة FOXC1 قد يشارك في ظهور الأعراض النفسية العصبية، مما يشير إلى أهمية المتابعة طويلة الأمد من منظور الصحة النفسية.
تم الإشارة إلى أن طفرات FOXC1 قد تحفز مرض الأوعية الدموية الدماغية الصغيرة وتزيد من خطر السكتة الدماغية 4). تعتبر الوقاية والتدخل المبكر لأمراض الأوعية الدموية العصبية لدى مرضى متلازمة أكسفيلد-ريجر من موضوعات البحث المستقبلية.
يتقدم فهم الآلية المرضية لـ “تصلب” القرنية الناجم عن طفرات FOXC1 1). من المتوقع أن يؤدي فهم الآليات الجزيئية لعتمة القرنية إلى تطوير علاجات جديدة باستخدام العلاج الجيني والأدوية المضادة للتليف والمواد الحيوية 1).