ARS प्रकार 1
कारण जीन: PITX2 (4q25)
मुख्य असामान्यताएं: पूर्वकाल नेत्र खंड असामान्यताएं, दांतों की असामान्यताएं, नाभि के आसपास अतिरिक्त त्वचा/नाभि हर्निया, कपाल-चेहरे की असामान्यताएं, हृदय संबंधी असामान्यताएं

एक्सेनफेल्ड-रीगर सिंड्रोम (ARS) पूर्वकाल खंड विकृति और प्रणालीगत असामान्यताओं का एक संयोजन है। मूल कारण तंत्रिका शिखा कोशिकाओं के प्रवास और विभेदन में असामान्यता माना जाता है। भ्रूण के अंतिम चरण में, पूर्वकाल कक्ष को ढकने वाली अविभेदित एंडोथेलियल कोशिकाओं का आइरिस और कोण से सामान्य रूप से गायब होना बाधित होता है, और उनका अवशेष रेशेदार बंधन और आइरिस के ऊंचे जुड़ाव का कारण बनता है।
ऐतिहासिक पृष्ठभूमि के रूप में, 1920 में एक्सेनफेल्ड ने पश्च भ्रूणीय वलय (श्वाल्बे रेखा का आगे की ओर विस्थापन और मोटा होना) और आइरिस प्रक्रियाओं का वर्णन किया। 1934-1935 में रीगर ने आइरिस हाइपोप्लासिया, प्यूपिलरी विचलन और पॉलीकोरिया की अतिरिक्त रिपोर्ट दी। वर्तमान में इसे तीन चरणों में वर्गीकृत किया गया है:
इन सबको मिलाकर एक्सेनफेल्ड-रीगर सिंड्रोम कहा जाता है। 50-60% मामलों में ग्लूकोमा होता है, यह ऑटोसोमल प्रभावी वंशानुक्रम है और आमतौर पर द्विपक्षीय होता है। मोतियाबिंद और लेंस की असामान्य स्थिति भी उच्च दर से जुड़ी होती है।
महामारी विज्ञान: प्रचलन लगभग 1/200,000 माना जाता था, लेकिन हाल की रिपोर्टों में 1/50,000 से 1/100,000 का अनुमान है2)4)। लिंग भेद नहीं है, और अक्सर शैशवावस्था में निदान किया जाता है।
आनुवंशिक प्रकार वर्गीकरण निम्नलिखित है:
ARS प्रकार 1
कारण जीन: PITX2 (4q25)
मुख्य असामान्यताएं: पूर्वकाल नेत्र खंड असामान्यताएं, दांतों की असामान्यताएं, नाभि के आसपास अतिरिक्त त्वचा/नाभि हर्निया, कपाल-चेहरे की असामान्यताएं, हृदय संबंधी असामान्यताएं
ARS प्रकार 2
कारण जीन: 13q14 (अनिश्चित)
मुख्य असामान्यताएं: पूर्वकाल नेत्र खंड असामान्यताएं, ग्लूकोमा। प्रकार 1 और 3 की तुलना में प्रणालीगत असामान्यताएं कम होती हैं।
ARS प्रकार 3
कारण जीन: FOXC1 (6p25)
मुख्य असामान्यताएं: पूर्वकाल नेत्र खंड असामान्यताएं, ग्लूकोमा, संवेदी श्रवण हानि, अलिंद सेप्टल दोष, गुर्दे की असामान्यताएं, श्वेत पदार्थ घाव
FOXC1 और PITX2 के उत्परिवर्तन ARS के 40-70% मामलों में होते हैं5)। हालांकि, ARS के 60% मामलों में कारण जीन अज्ञात है4), और आनुवंशिक विविधता बहुत अधिक है।
बाल्यावस्था/युवावस्था में शुरू होने वाले ग्लूकोमा के बड़े रजिस्ट्री विश्लेषण में, आणविक निदान दर 56.5% थी11)। FOXC1 उत्परिवर्तन 20.3%, PITX2 उत्परिवर्तन 17.4%, और PAX6 उत्परिवर्तन 10.1% थे, और ज्ञात जीनों द्वारा स्पष्ट नहीं किए जा सकने वाले मामले भी कम नहीं हैं11)।
इन्हें कारण जीन द्वारा अलग किया जाता है। प्रकार 1 PITX2 (4q25) उत्परिवर्तन के कारण होता है और इसमें दांत, नाभि और चेहरे की हड्डियों की असामान्यताएं होती हैं। प्रकार 3 FOXC1 (6p25) उत्परिवर्तन के कारण होता है और इसमें बहरापन, हृदय दोष, गुर्दे की असामान्यताएं और तंत्रिका संबंधी असामान्यताएं होती हैं। प्रकार 2 13q14 पर स्थित है लेकिन कारण जीन अनिश्चित है, और इसमें मुख्य रूप से पूर्वकाल नेत्र खंड असामान्यताएं और ग्लूकोमा होता है। आनुवंशिक परीक्षण द्वारा निश्चित निदान किया जा सकता है।
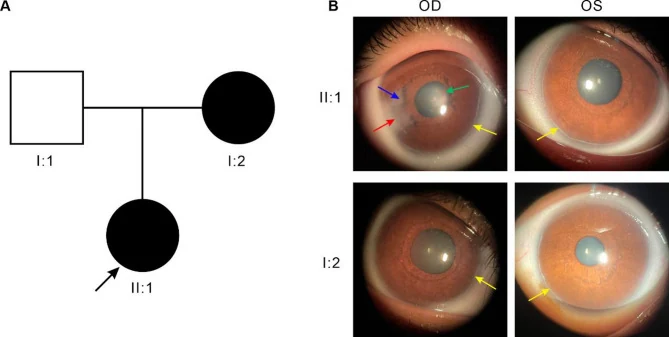
प्रमुख नेत्र निष्कर्ष नीचे दिए गए हैं।
| नेत्र निष्कर्ष | विशेषताएँ |
|---|---|
| पश्च भ्रूणीय वलय | श्वाल्बे रेखा का पूर्ववर्ती विस्थापन और मोटा होना |
| आइरिस प्रक्रियाएँ | पतले धागे जैसी से चौड़ी पट्टी जैसी |
| पुतली का विचलन | पश्च भ्रूणीय वलय के विपरीत दिशा में विचलन |
| छद्म बहुपुतली | परितारिका स्ट्रोमा का छिद्र-जैसा दिखना |
| यूवीय बहिर्वर्तन | परितारिका वर्णक उपकला का उलटना |
पश्च भ्रूणीय वलय श्वाल्बे रेखा पर अविभेदित कोशिकाओं का अवशेष है, जो लिंबस से 0.5-2.0 मिमी केंद्र की ओर लिंबस के साथ एक रेखा के रूप में दिखाई देता है। यह अक्सर पूर्ण परिधि में नहीं बल्कि एक भाग में सीमित होता है। यदि उभरी हुई श्वाल्बे रेखा और परितारिका आपस में चिपक जाती है, तो इसे एक्सेनफेल्ड असामान्यता कहा जाता है, और यदि परितारिका स्ट्रोमा का शोष भी हो, तो इसे रीगर असामान्यता कहा जाता है।
कॉर्निया सामान्यतः पारदर्शी होता है और एंडोथेलियम सामान्य होता है, लेकिन अवशिष्ट ऊतक के साथ भौतिक संपर्क से द्वितीयक रूप से कॉर्नियल धुंधलापन हो सकता है। कॉर्नियल धुंधलापन अक्सर परिधीय भाग तक सीमित होता है और आमतौर पर दृष्टि को सीधे प्रभावित नहीं करता। हालांकि, FOXC1 उत्परिवर्तन में कॉर्नियल धुंधलापन और कॉर्नियल नववाहिकीकरण अधिक स्पष्ट होता है, और PITX2 उत्परिवर्तन की तुलना में कॉर्नियल असामान्यता की गंभीरता अधिक होती है तथा ग्लूकोमा की आवृत्ति भी अधिक होती है1)।
कोण निष्कर्षों में परितारिका का ऊंचा जुड़ाव, डोरी जैसे यूवीय अवशेष, और श्वाल्बे रेखा का मोटा होना (पश्च भ्रूणीय वलय) शामिल हैं। माइक्रोस्फेरोफेकिया और लेंस सब्लक्सेशन के सह-अस्तित्व के मामले भी रिपोर्ट किए गए हैं7)।
ग्लूकोमा 50-60% मामलों में सह-अस्तित्व में होता है। शैशवावस्था से ही अंतर्नेत्र दबाव बढ़ सकता है, लेकिन अधिकांश मामले बचपन से युवावस्था में शुरू होते हैं। कुछ मामलों में प्रगतिशील दृष्टि हानि के कारण निदान होता है; पूर्व खंड निष्कर्षों और ग्लूकोमेटस परिवर्तनों को अनदेखा न करना महत्वपूर्ण है6)।
Li et al. (2021) के 7 वर्षीय लड़के (ARS प्रकार 3, de novo FOXC1 उत्परिवर्तन) में कॉर्नियल व्यास 14 मिमी, अक्षीय लंबाई 27.16/26.56 मिमी, C/D अनुपात 0.9, IOP 33/20 mmHg था। जन्म के 36वें दिन से दोनों आँखों में एंटीग्लूकोमा सर्जरी की आवश्यकता थी5)।
प्रणालीगत निष्कर्ष निम्नलिखित हैं:
लगभग 50-60% ARS रोगियों में ग्लूकोमा विकसित होता है। यह अक्सर बचपन या युवा वयस्कता में शुरू होता है, लेकिन शैशवावस्था में भी अंतर्गर्भाशयी दबाव बढ़ सकता है। नियमित अंतर्गर्भाशयी दबाव माप और ऑप्टिक तंत्रिका मूल्यांकन आवश्यक है। अधिक जानकारी के लिए, “मानक उपचार” अनुभाग देखें।
ARS एक ऑटोसोमल प्रभावी विकार है जिसमें पूर्ण पैठ होती है। हालांकि, एक ही परिवार में समान आनुवंशिक उत्परिवर्तन वाले व्यक्तियों में नैदानिक अभिव्यक्तियाँ बहुत भिन्न हो सकती हैं (परिवर्तनशील अभिव्यक्ति)1)।
बड़े पैमाने के कोहोर्ट अध्ययन में, FOXC1 और PITX2 उत्परिवर्तन बचपन से वयस्कता तक ग्लूकोमा के व्यापक स्पेक्ट्रम से जुड़े थे9)। नैदानिक रूप से प्राथमिक जन्मजात ग्लूकोमा (PCG) के रूप में प्रारंभिक निदान वाले मामलों को आनुवंशिक परीक्षण द्वारा पुनर्वर्गीकृत किया जा सकता है। जब शिशुओं में पूर्व खंड के लक्षण सूक्ष्म होते हैं, तो आनुवंशिक परीक्षण सटीक रोग प्रकार निदान में योगदान कर सकता है।
PITX2 जीन के आसपास सूक्ष्म विलोपन वाले मामलों में, NEUROG2, UGT8 और NDST4 के विलोपन के ओवरलैप होने से विकासात्मक विलंब और बौद्धिक अक्षमता हो सकती है8)3)।
Kawanami et al. (2023) ने 4q25 पर 2.5 Mb के सूक्ष्म विलोपन (PITX2, NEUROG2 और ANK2 सहित) वाले 3 वर्षीय जापानी लड़के की रिपोर्ट दी। उसे नाभि हर्निया, आइरिस कोलोबोमा और विकासात्मक विलंब था, लेकिन ANK2 विलोपन के बावजूद ईसीजी सामान्य था। NEUROG2 की हैप्लोइन्सफिशिएंसी को विकासात्मक विलंब का संभावित कारण माना गया8)।
ऑटोसोमल प्रभावी वंशानुक्रम के कारण, उत्परिवर्तन वाले माता-पिता से संचरण की संभावना 50% है। पैठ पूर्ण है, लेकिन फेनोटाइप में व्यक्तिगत भिन्नता होती है: एक ही उत्परिवर्तन के साथ लक्षणों की गंभीरता काफी भिन्न हो सकती है1)। आनुवंशिक परीक्षण और आनुवंशिक परामर्श की सिफारिश की जाती है।
ARS का निदान द्विपक्षीय कोण असामान्यताओं और आइरिस असामान्यताओं पर आधारित है। निदान की शर्त पोस्टीरियर एम्ब्रियोटॉक्सन के साथ परिधीय आइरिस का आंशिक जुड़ाव है13)। यदि स्लिट लैंप से पोस्टीरियर एम्ब्रियोटॉक्सन की पुष्टि नहीं होती है, तो गोनियोस्कोपी आवश्यक है। प्रणालीगत असामान्यताओं के मामले में, ARS सिंड्रोम के रूप में बाल रोग में पूर्ण जांच का अनुरोध किया जाता है13)।
ध्यान दें कि सामान्य जनसंख्या के 8-15% में हल्का पोस्टीरियर एम्ब्रियोटॉक्सन देखा जाता है, लेकिन यह अकेले ग्लूकोमा आदि से जुड़ा नहीं है। निदान में पारिवारिक इतिहास लेना भी महत्वपूर्ण है।
ग्लूकोमा उपचार दिशानिर्देश (5वें संस्करण) में बाल चिकित्सा ग्लूकोमा के वर्गीकरण में, ARS को जन्मजात नेत्र विकास संबंधी असामान्यता से जुड़े ग्लूकोमा के विशिष्ट उदाहरण के रूप में रखा गया है13)। जन्म से मौजूद नेत्र विकास संबंधी असामान्यताएं बाल चिकित्सा ग्लूकोमा के निदान मानदंडों को पूरा करने पर निदान किया जाता है।
ARS से विभेदित की जाने वाली प्रमुख बीमारियाँ नीचे दी गई हैं।
| रोग | ARS से अंतर |
|---|---|
| ICE सिंड्रोम | एकतरफा, अधिग्रहित, महिला प्रधान |
| पीटर्स असामान्यता | केंद्रीय कॉर्नियल अपारदर्शिता, डेसीमेट झिल्ली की कमी |
| एनिरिडिया | कॉर्नियल पैन्नस, फोवियल हाइपोप्लासिया |
| पोस्टीरियर पॉलीमॉर्फस कॉर्नियल डिस्ट्रोफी | द्विपक्षीय, पारिवारिक, लिंग भेद नहीं |
ICE सिंड्रोम (प्रगतिशील आइरिस शोष, चैंडलर सिंड्रोम आदि) का ARS से विभेदन महत्वपूर्ण है, लेकिन सबसे बड़ा विभेदक बिंदु यह है कि ICE एकतरफा और अधिग्रहित है, जबकि ARS द्विपक्षीय और जन्मजात है।
ARS का कोई कारणात्मक उपचार वर्तमान में मौजूद नहीं है। उपचार का केंद्र ग्लूकोमा प्रबंधन और प्रणालीगत जटिलताओं की निगरानी है। उपचार रणनीति प्रारंभिक-शुरुआत विकासात्मक ग्लूकोमा (प्राथमिक जन्मजात ग्लूकोमा: PCG) के समान है 13)।
ग्लूकोमा लगभग 50-60% ARS मामलों में जटिलता के रूप में होता है। दवा उपचार सामान्य ग्लूकोमा के अनुसार किया जाता है, लेकिन अक्सर अप्रभावी होता है।
जलीय हास्य उत्पादन अवरोधक
बीटा-ब्लॉकर्स : पहली पंक्ति के विकल्पों में से एक। सुरक्षित और प्रभावी, लेकिन बच्चों में अक्सर अप्रभावी।
कार्बोनिक एनहाइड्रेज़ अवरोधक (CAI) आई ड्रॉप : ब्रिन्ज़ोलामाइड आदि। बीटा-ब्लॉकर्स के साथ संयोजन संभव।
अल्फा-2 एगोनिस्ट (ब्रिमोनिडाइन) : 2 वर्ष से कम उम्र में न्यूरोसाइकिएट्रिक प्रभावों (एपनिया, ब्रैडीकार्डिया, हाइपोटेंशन, मांसपेशी हाइपोटोनिया, सीएनएस अवसाद) के कारण वर्जित है 13)।
जलीय हास्य बहाव बढ़ाने वाली दवाएं
प्रोस्टाग्लैंडीन एनालॉग्स : लैटानोप्रोस्ट, ट्रैवोप्रोस्ट आदि। बच्चों में प्रभाव वयस्कों की तुलना में कमजोर माना जाता है 13)।
उदाहरण : 7 वर्षीय लड़के में ट्रैवोप्रोस्ट + ब्रिन्ज़ोलामाइड से दीर्घकालिक नियंत्रण 5)। 77 वर्षीय पुरुष में लैटानोप्रोस्ट, टिमोलोल और ब्रिन्ज़ोलामाइड के बावजूद IOP 35 mmHg पर नियंत्रण कठिन था 2)।
एक रिपोर्ट के अनुसार प्रोस्टेनॉइड FP रिसेप्टर एगोनिस्ट और बीटा-ब्लॉकर्स के बीच प्रभाव में कोई अंतर नहीं है 13)।
शिशुओं में, शरीर के वजन और सतह क्षेत्र के सापेक्ष आई ड्रॉप की खुराक अपेक्षाकृत अधिक होती है, इसलिए जहां तक संभव हो कम सांद्रता वाली दवाओं का उपयोग किया जाना चाहिए 13)।
यदि दवा उपचार से अंतःनेत्र दबाव नियंत्रित नहीं होता है, तो शल्य चिकित्सा की जाती है 10)13)।
GDD के बाद जटिलताओं में रिपोर्ट की गई हैं: उथला पूर्वकाल कक्ष 13.6%, हाइपोटोनी 11.7%, कोरॉइडल इफ्यूजन 8.3%, एंडोफ्थैल्मिटिस 1.7% 14)।
Chakraborty et al. (2022) ने Axenfeld-Rieger सिंड्रोम से जुड़े रेटिना डिटेचमेंट (15 वर्षीय लड़का) का एक मामला रिपोर्ट किया। इसमें माइक्रोस्फेरोफेकिया और लेंस सब्लक्सेशन था; विट्रेक्टॉमी के बाद IOP बढ़कर 41 mmHg हो गया और स्क्लेरल स्टेफिलोमा बन गया। डायोड सिलिअरी फोटोकोएग्यूलेशन किया गया, अंततः IOP 18 mmHg प्राप्त हुआ 7)।
कोण सर्जरी की सफलता दर PCG से कम है 13)। MMC के साथ ट्रैबेक्यूलेक्टोमी में 2 वर्ष की दीर्घकालिक सफलता दर लगभग 59% है, GDD में 12 महीने में 87% और 24 महीने में 77% बताई गई है 14)। दुर्दम्य मामलों में कई बार सर्जरी की आवश्यकता हो सकती है।
ARS का मूल कारण तंत्रिका शिखा कोशिकाओं के प्रवास और विभेदन में दोष है। पूर्वकाल कक्ष, पूर्वकाल खंड कोण, चेहरे की हड्डियों, दांतों, हृदय प्रणाली और नाभि के आसपास की त्वचा में तंत्रिका शिखा कोशिकाओं के विकास में बाधा के कारण बहु-अंग विकृति उत्पन्न होती है।
भ्रूण के अंतिम चरण में, पूर्वकाल कक्ष को ढकने वाली अविभेदित एंडोथेलियल कोशिकाएं सामान्य रूप से परितारिका और कोण से गायब हो जाती हैं। ARS में यह गायब होने की प्रक्रिया बाधित होती है, और अविभेदित एंडोथेलियल कोशिकाएं परितारिका पर बनी रहती हैं, जिससे रज्जु जैसी संरचनाएं बनती हैं। कोण में, परितारिका का उच्च जुड़ाव होता है, जो ट्रैबेकुलर मेशवर्क को यांत्रिक रूप से ढक देता है।
ऊतकीय रूप से, कॉर्निया की पिछली सतह से पूर्वकाल कक्ष, कोण और परितारिका की सतह तक डेसीमेट झिल्ली जैसी झिल्ली के साथ एंडोथेलियल कोशिकाओं की एक असामान्य एकल परत फैली होती है। यूवियल एक्ट्रोपियन और प्यूपिलरी विचलन वाले चतुर्थांशों में झिल्ली मौजूद होती है, जबकि विपरीत चतुर्थांशों में परितारिका शोष देखा जाता है।
FOXC1 और PITX2 दोनों प्रतिलेखन कारक हैं, जो विशिष्ट DNA अनुक्रमों से जुड़कर अधोप्रवाह जीनों की अभिव्यक्ति को नियंत्रित करते हैं। दोनों पूर्वकाल खंड विकास में सहक्रियात्मक रूप से कार्य करते हैं और सामान्य अधोप्रवाह लक्ष्य जीनों को नियंत्रित करते हैं 3)। FOXC1 का फोर्कहेड डोमेन (110 अमीनो एसिड का DNA-बाइंडिंग डोमेन) कार्यात्मक रूप से सबसे महत्वपूर्ण है 2), और इस डोमेन के उत्परिवर्तन न्यूरोसाइकियाट्रिक लक्षणों से अधिक मजबूती से जुड़े होने का सुझाव दिया गया है।
अंतर्गर्भाशयी दबाव बढ़ने के दो तंत्र बताए गए हैं।
आइरिस की अनुपस्थिति की मात्रा और कोण में आइरिस प्रक्रियाओं की संख्या ग्लूकोमा की गंभीरता से आवश्यक रूप से संबंधित नहीं होती है। हालांकि, कोण में आइरिस आसंजन की उच्च डिग्री ग्लूकोमा की संभावना को बढ़ाती है।
FOXC1 उत्परिवर्तन अन्य उत्परिवर्तनों की तुलना में जन्मजात ग्लूकोमा को अधिक बढ़ावा देता है 1), और सिलिअरी बॉडी और जल निकासी कोण की रूपात्मक असामान्यताएं IOP वृद्धि में योगदान कर सकती हैं 1)।
FOXC1 उत्परिवर्ती चूहों में, कॉर्नियल स्ट्रोमा में कोलेजन फाइबर की कमी और संरचनात्मक असामान्यताएं, साथ ही कॉर्नियल स्ट्रोमल कोशिकाओं की क्षति देखी जाती है 1)। इसके अलावा, FOXC1 कॉर्नियल नववाहिकीकरण के अवरोधक के रूप में कार्य करता है (VEGF की जैवउपलब्धता के नियंत्रण के माध्यम से) 1); FOXC1 उत्परिवर्तन के कारण इस अवरोध के खत्म होने से कॉर्नियल नववाहिकीकरण होता है।
FOXC1, FOX परिवार के प्रतिलेखन कारक के रूप में, मस्तिष्क के विकास में भी महत्वपूर्ण भूमिका निभाता है 4)।
एक व्यवस्थित समीक्षा में बताया गया कि ARS के 41.3% मामलों में श्वेत पदार्थ की असामान्यताएं दिखाई देती हैं 4)। FOXC1 उत्परिवर्तन मस्तिष्क छोटे वाहिका रोग (CSVD), श्वेत पदार्थ हाइपरइंटेंसिटी, बढ़े हुए पेरिवास्कुलर स्पेस, माइक्रोहेमरेज और लैकुनर इन्फार्क्ट को प्रेरित कर सकता है।
Ohkubo et al. (2025) ने एक 2 वर्षीय जापानी लड़के (FOXC1 उत्परिवर्तन: c.240del, p.Y81Ifs21) में मस्तिष्क MRI द्वारा पेरिवेंट्रिकुलर श्वेत पदार्थ घाव, बढ़े हुए पेरिवास्कुलर स्पेस और वर्टेब्रोबैसिलर धमनी की टेढ़ापन-फैलाव की पुष्टि की। पिता को 18 वर्ष की आयु में मस्तिष्क रोधगलन का इतिहास था 4)।
FOXC1 उत्परिवर्तन के 95 मामलों की समीक्षा में 6.3% में न्यूरोसाइकियाट्रिक लक्षण (सीखने में कठिनाई, मिर्गी, बौद्धिक अक्षमता, ईर्ष्या भ्रम, आदि) पाए गए, और फोर्कहेड डोमेन उत्परिवर्तन वाले 83.3% मामलों में न्यूरोसाइकियाट्रिक लक्षण दिखाई दिए 2)।
हाँ। FOXC1 उत्परिवर्तन वाले ARS में लगभग 41% मामलों में श्वेत पदार्थ की असामान्यताएं दिखाई देती हैं, ऐसा एक व्यवस्थित समीक्षा में बताया गया है 4)। FOXC1 उत्परिवर्तन मस्तिष्क छोटे वाहिका रोग और स्ट्रोक के जोखिम से जुड़ा हो सकता है; विशेष रूप से FOXC1 उत्परिवर्तन प्रकार के ARS में दीर्घकालिक तंत्रिका संबंधी अनुवर्ती महत्वपूर्ण है।
हाल के वर्षों में, अगली पीढ़ी अनुक्रमण और संपूर्ण जीनोम अनुक्रमण के माध्यम से कई नए उत्परिवर्तनों की सूचना दी गई है।
Wowra et al. (2024) ने तीन पोलिश बहनों में ARS में FOXC1 एक्सॉन 1 के एक भाग और संपूर्ण 3’UTR को शामिल करते हुए एक बड़े विलोपन (नया उत्परिवर्तन) की पहचान की। एक ही परिवार में भी फेनोटाइप काफी भिन्न थे, और शुरू में चैंडलर सिंड्रोम का गलत निदान किया गया था 1).
Jiang et al. (2024) ने एक चीनी परिवार में ARS प्रकार 1 में PITX2 युक्त 6.15 Mb गुणसूत्र 4q25 विलोपन, 45.71 Mb व्युत्क्रमण और 14 bp विलोपन सहित एक जटिल जीनोमिक पुनर्व्यवस्था की पहचान की। एक 11 वर्षीय लड़की में IOP 43.5/44.0 mmHg था 3).
अन्य नए उत्परिवर्तन रिपोर्टों में FOXC1 p.Phe136Leu (फोर्कहेड डोमेन) 2), FOXC1 p.S82R (डी नोवो उत्परिवर्तन) 5), और FOXC1 c.240del, p.Y81Ifs21 4) शामिल हैं।
Yoshino et al. (2024) ने ARS प्रकार 3 के एक 77 वर्षीय जापानी पुरुष का मामला बताया। 72 वर्ष की आयु से ईर्ष्या संबंधी भ्रम प्रकट हुए, और ल्यूकोएन्सेफैलोपैथी की पुष्टि हुई। FOXC1 उत्परिवर्तन वाले 95 मामलों की साहित्य समीक्षा में 6.3% (6/95) में न्यूरोसाइकियाट्रिक लक्षण पाए गए, जिनमें से 83.3% (5/6) में फोर्कहेड डोमेन उत्परिवर्तन था 2).
यह निष्कर्ष बताता है कि FOXC1 उत्परिवर्तन का कार्यात्मक डोमेन न्यूरोसाइकियाट्रिक लक्षणों की अभिव्यक्ति में शामिल हो सकता है, जो मानसिक स्वास्थ्य के दृष्टिकोण से दीर्घकालिक अनुवर्ती के महत्व को इंगित करता है।
यह बताया गया है कि FOXC1 उत्परिवर्तन CSVD को प्रेरित कर सकते हैं और स्ट्रोक के जोखिम को बढ़ा सकते हैं 4)। ARS रोगियों में न्यूरोवास्कुलर रोगों की रोकथाम और प्रारंभिक हस्तक्षेप का महत्व भविष्य के शोध विषयों के रूप में माना जाता है।
FOXC1 उत्परिवर्तन के कारण कॉर्नियल “स्क्लेरलाइज़ेशन” की रोगविज्ञान की समझ बढ़ रही है 1)। कॉर्नियल अपारदर्शिता के आणविक तंत्र की समझ से जीन थेरेपी, एंटी-फाइब्रोटिक दवाओं और बायोमटेरियल का उपयोग करके नए उपचारों के विकास की उम्मीद है 1).