ARS tipo 1
Gen causante: PITX2 (4q25)
Anomalías principales: Anomalías del segmento anterior, anomalías dentales, piel periumbilical redundante/hernia umbilical, anomalías craneofaciales, anomalías cardiovasculares

El síndrome de Axenfeld-Rieger (ARS) es un grupo de trastornos congénitos que combinan displasia del segmento anterior y anomalías sistémicas. Se cree que la etiología fundamental es una migración y diferenciación anormal de las células de la cresta neural. El proceso normal de desaparición de las células endoteliales indiferenciadas que recubren la cámara anterior desde el iris y el ángulo al final de la vida fetal se ve alterado, y sus restos causan formación de bandas e inserción alta del iris.
Antecedentes históricos: En 1920, Axenfeld describió el embriotoxón posterior (desplazamiento anterior y engrosamiento de la línea de Schwalbe) y procesos del iris. En 1934-1935, Rieger agregó informes de hipoplasia del iris, corectopia y policoria. Actualmente se clasifica en las siguientes tres etapas:
Estos se denominan colectivamente síndrome de Axenfeld-Rieger. El glaucoma ocurre en el 50-60% de los casos, y generalmente es bilateral con herencia autosómica dominante. La catarata y la luxación del cristalino también se asocian con frecuencia.
Epidemiología: La prevalencia se ha reportado como aproximadamente 1/200,000, pero estimaciones recientes sugieren 1/50,000 a 100,000 2)4). No hay predilección por sexo y a menudo se diagnostica en la infancia o la primera infancia.
Clasificación genética es la siguiente:
ARS tipo 1
Gen causante: PITX2 (4q25)
Anomalías principales: Anomalías del segmento anterior, anomalías dentales, piel periumbilical redundante/hernia umbilical, anomalías craneofaciales, anomalías cardiovasculares
ARS tipo 2
Gen causante: 13q14 (no identificado)
Anomalías principales: Anomalías del segmento anterior, glaucoma. Las anomalías sistémicas son menos frecuentes que en los tipos 1 y 3
ARS tipo 3
Gen causante: FOXC1 (6p25)
Anomalías principales: Anomalías del segmento anterior, glaucoma, hipoacusia neurosensorial, defecto del tabique interauricular, anomalías renales, lesiones de la sustancia blanca
Las mutaciones en FOXC1 y PITX2 representan del 40 al 70% de los casos de ARS 5). Sin embargo, en el 60% de los casos de ARS el gen causante no se ha identificado 4), lo que indica una heterogeneidad genética considerable.
En un análisis de un registro a gran escala de glaucoma de inicio en la infancia y en adultos jóvenes, la tasa de diagnóstico molecular fue del 56,5% 11). Las mutaciones en FOXC1 representaron el 20,3%, las mutaciones en PITX2 el 17,4% y las mutaciones en PAX6 el 10,1%; un número considerable de casos no pudo explicarse por genes conocidos 11).
Se distinguen por el gen causante. El tipo 1 es causado por mutaciones en PITX2 (4q25) y se asocia con anomalías dentales, umbilicales y de los huesos faciales. El tipo 3 es causado por mutaciones en FOXC1 (6p25) y se asocia con pérdida auditiva, defectos cardíacos, anomalías renales y anomalías neurológicas. El tipo 2 se mapea en 13q14 pero el gen causante aún no se ha identificado; se presenta principalmente con anomalías del segmento anterior y glaucoma. Las pruebas genéticas pueden proporcionar un diagnóstico definitivo.
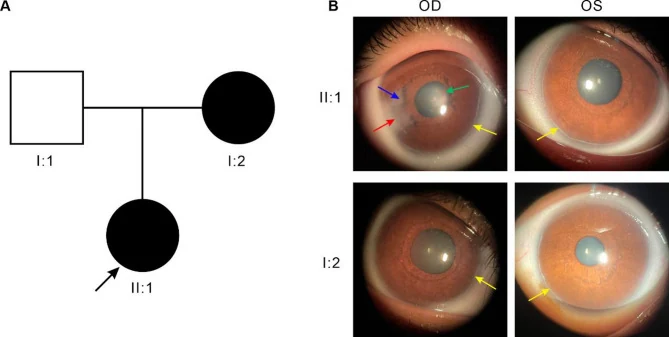
Principales hallazgos oculares se muestran a continuación.
| Hallazgo ocular | Características |
|---|---|
| Embriotóxon posterior | Desplazamiento anterior y engrosamiento de la línea de Schwalbe |
| Procesos del iris | Desde filiformes hasta bandas anchas |
| Desviación pupilar | Desviación en dirección opuesta al embriotoxón posterior |
| Pseudopolicoria | Aspecto de perforación del estroma del iris |
| Ectropión uveal | Eversión del epitelio pigmentario del iris |
El embriotoxón posterior es un remanente de células indiferenciadas en la línea de Schwalbe, observado linealmente a lo largo del limbo, 0.5–2.0 mm hacia el centro desde el limbo. A menudo es localizado en lugar de circunferencial. La adhesión entre la línea de Schwalbe prominente y el iris se denomina anomalía de Axenfeld, y si se acompaña de atrofia del estroma del iris, se denomina anomalía de Rieger.
La córnea suele ser transparente con estructura endotelial normal, pero puede ocurrir opacidad corneal secundaria debido al contacto físico con el tejido residual. La opacidad corneal a menudo se localiza en la periferia y generalmente no afecta directamente la visión. Sin embargo, en las mutaciones de FOXC1, la opacidad corneal y la neovascularización corneal son más prominentes, y el grado de anomalía corneal es mayor y la frecuencia de glaucoma es más alta en comparación con las mutaciones de PITX2 1).
Los hallazgos gonioscópicos incluyen inserción alta del iris, restos de membrana pupilar persistente y engrosamiento de la línea de Schwalbe (embriotoxón posterior). También se han reportado casos complicados con esferofaquia o subluxación del cristalino 7).
El glaucoma ocurre en el 50–60% de los casos. La elevación de la presión intraocular puede ocurrir en la infancia, pero la mayoría de los casos se desarrollan en la niñez o la juventud. Algunos casos se diagnostican después de una pérdida progresiva de la visión, por lo que es importante no pasar por alto los hallazgos del segmento anterior y los cambios glaucomatosos 6).
Li et al. (2021) reportaron un niño de 7 años (ARS tipo 3, mutación de novo FOXC1) con diámetro corneal de 14 mm, longitud axial de 27.16/26.56 mm, relación copa-disco de 0.9 y PIO de 33/20 mmHg. Se requirió cirugía antiglaucomatosa en ambos ojos desde los 36 días de vida 5).
Los hallazgos sistémicos son los siguientes:
Alrededor del 50–60% de los casos de ARS presentan glaucoma. A menudo se desarrolla durante la infancia hasta la juventud, pero también hay casos que presentan elevación de la presión intraocular desde la lactancia. Es necesaria la medición regular de la presión intraocular y la evaluación del nervio óptico. Para más detalles, consulte la sección “Tratamiento estándar”.
El ARS muestra herencia autosómica dominante con penetrancia completa. Sin embargo, incluso dentro de la misma familia portadora de la misma mutación genética, existe una gran variabilidad en la presentación clínica (expresividad variable)1).
Estudios de cohortes grandes han demostrado que las mutaciones en FOXC1 y PITX2 se asocian con un amplio espectro de glaucoma desde la infancia hasta la edad adulta9). Los casos diagnosticados inicialmente como PCG (glaucoma congénito primario) pueden ser reclasificados mediante pruebas genéticas, y las pruebas genéticas contribuyen al diagnóstico preciso cuando los hallazgos del segmento anterior en lactantes son sutiles.
En casos con microdeleciones alrededor del gen PITX2, las deleciones superpuestas de NEUROG2, UGT8 y NDST4 pueden causar retraso del desarrollo y discapacidad intelectual 8)3).
Kawanami et al. (2023) reportaron un niño japonés de 3 años con una microdeleción de 2.5 Mb en 4q25 (que incluye PITX2, NEUROG2 y ANK2). Presentó onfalocele, coloboma de iris y retraso del desarrollo, pero su electrocardiograma fue normal a pesar de la deleción de ANK2. La haploinsuficiencia de NEUROG2 se consideró una causa candidata del retraso del desarrollo 8).
Debido a la herencia autosómica dominante, la probabilidad de transmisión de un padre afectado es del 50%. La penetrancia es completa, pero el fenotipo varía; incluso con la misma mutación, la gravedad de los síntomas puede diferir mucho 1). Se recomiendan pruebas genéticas y asesoramiento genético.
El diagnóstico del ARS se basa en anomalías del ángulo e iris bilaterales. La adhesión parcial del iris periférico al embriotoxón posterior se considera un criterio diagnóstico 13). Si el embriotoxón posterior no es visible en la lámpara de hendidura, es necesaria la gonioscopia. Si hay anomalías sistémicas, se deriva a pediatría para una evaluación sistémica completa como síndrome de ARS 13).
Tenga en cuenta que un embriotoxón posterior leve se observa en el 8–15% de la población normal, pero por sí solo no se asocia con glaucoma. La historia familiar también es importante en el diagnóstico.
En el sistema de clasificación del glaucoma infantil de las Guías de Práctica Clínica de Glaucoma (5ª edición), el ARS se posiciona como un ejemplo representativo de glaucoma asociado con anomalías oculares congénitas 13). Se diagnostica cuando las anomalías oculares presentes al nacer cumplen los criterios diagnósticos de glaucoma infantil.
Se presentan las principales enfermedades que deben diferenciarse del ARS.
| Enfermedad | Diferencias con ARS |
|---|---|
| Síndrome ICE | Unilateral, adquirido, predominio femenino |
| Anomalía de Peters | Opacidad corneal central, defecto de la membrana de Descemet |
| Aniridia | Paño corneal, hipoplasia foveal |
| Distrofia corneal polimorfa posterior | Bilateral, familiar, sin diferencia de sexo |
El diagnóstico diferencial con el síndrome ICE (atrofia progresiva del iris, síndrome de Chandler, etc.) es importante, pero el punto clave de diferenciación es que el ICE es unilateral y adquirido, mientras que el ARS es bilateral y congénito.
Actualmente no existe un tratamiento curativo para el ARS en sí; el manejo se centra en el control del glaucoma y la vigilancia de complicaciones sistémicas. La estrategia de tratamiento sigue la del glaucoma de desarrollo de inicio temprano (glaucoma congénito primario: PCG) 13).
El glaucoma ocurre en aproximadamente el 50–60% de los casos de ARS. El tratamiento farmacológico sigue el del glaucoma general, pero a menudo es ineficaz.
Supresores de la producción de humor acuoso
Betabloqueantes: Una de las opciones de primera línea. Seguros y efectivos, pero a menudo ineficaces en niños.
Inhibidores de la anhidrasa carbónica (IAC) en colirio: p. ej., brinzolamida. Pueden combinarse con betabloqueantes.
Agonistas alfa-2 (brimonidina): Contraindicados en menores de 2 años debido a efectos neuropsiquiátricos (apnea, bradicardia, hipotensión, hipotonía muscular, depresión del SNC) 13).
Promotores del flujo de salida del humor acuoso
Análogos de prostaglandinas: p. ej., latanoprost, travoprost. La eficacia en niños se considera más débil que en adultos 13).
Ejemplo: Un niño de 7 años fue manejado a largo plazo con travoprost + brinzolamida 5). Un hombre de 77 años presentó PIO de 35 mmHg a pesar de latanoprost, timolol y brinzolamida, indicando control deficiente 2).
Hay un informe que no encontró diferencias en la eficacia entre los agonistas del receptor FP de prostanoides y los betabloqueantes 13).
En lactantes, la dosis de colirio es relativamente alta en comparación con el peso y la superficie corporal; por lo tanto, se debe usar la concentración más baja posible 13).
Si la presión intraocular no se puede controlar con medicamentos, se realiza cirugía10)13).
Las complicaciones después de la cirugía de GDD incluyen cámara anterior poco profunda 13.6%, hipotonía 11.7%, derrame coroideo 8.3% y endoftalmitis 1.7%14).
Chakraborty et al. (2022) reportaron un caso de desprendimiento de retina asociado a ARS (niño de 15 años). El paciente presentaba microesferofaquia y subluxación del cristalino; después de la vitrectomía, la PIO aumentó a 41 mmHg y se formó un estafiloma escleral. Se realizó ciclofotocoagulación con diodo, logrando finalmente una PIO de 18 mmHg7).
La tasa de éxito de la cirugía de ángulo es menor que para PCG13). Para la trabeculectomía con MMC, la tasa de éxito a largo plazo a 2 años es aproximadamente del 59%; para GDD, se reporta un 87% a los 12 meses y un 77% a los 24 meses14). Los casos refractarios pueden requerir múltiples cirugías.
La etiología fundamental del ARS es un defecto en la migración y diferenciación de las células de la cresta neural. El desarrollo alterado de las células de la cresta neural en la cámara anterior, el ángulo del segmento anterior, los huesos faciales, los dientes, el sistema cardiovascular y la piel periumbilical conduce a malformaciones multiorgánicas.
En la etapa embrionaria tardía, las células endoteliales indiferenciadas que cubren la cámara anterior normalmente desaparecen del iris y el ángulo. En ARS, este proceso de desaparición se ve afectado y las células endoteliales indiferenciadas permanecen en el iris, causando la formación de hebras. En el ángulo, se produce una inserción alta del iris, cubriendo mecánicamente la malla trabecular.
Histológicamente, una monocapa de células similares a endoteliales con una membrana similar a la de Descemet se extiende anormalmente desde la córnea posterior hasta la cámara anterior, el ángulo y la superficie del iris. La membrana está presente en los cuadrantes con ectropión uveal y corectopia, mientras que se observa atrofia del iris en los cuadrantes opuestos.
FOXC1 y PITX2 son ambos factores de transcripción que se unen a secuencias de ADN específicas y regulan la expresión de genes下游. Actúan sinérgicamente en el desarrollo del segmento anterior y regulan genes diana comunes3). El dominio forkhead (dominio de unión al ADN de 110 aminoácidos) de FOXC1 es funcionalmente el más importante2), y se sugiere que las mutaciones en este dominio se asocian más fuertemente con síntomas neuropsiquiátricos.
Se han señalado los siguientes dos mecanismos para la elevación de la presión intraocular.
El grado de defecto del iris y la cantidad de procesos iridianos en el ángulo no se correlacionan necesariamente con la gravedad del glaucoma. Sin embargo, un grado extenso de sinequias anteriores periféricas en el ángulo predispone al glaucoma.
Las mutaciones de FOXC1 promueven el glaucoma congénito más que otras mutaciones 1), y las anomalías morfológicas del cuerpo ciliar y del ángulo de drenaje pueden contribuir a la elevación de la PIO 1).
En ratones con mutación de FOXC1, se observan reducción de fibras de colágeno y anomalías estructurales en el estroma corneal, así como daño a los queratocitos 1). Además, FOXC1 funciona como un supresor de la angiogénesis corneal (mediante la regulación de la biodisponibilidad del VEGF) 1), y la pérdida de esta supresión debido a la mutación de FOXC1 conduce a neovascularización corneal.
FOXC1, como factor de transcripción de la familia FOX, también desempeña un papel importante en el desarrollo cerebral 4).
Una revisión sistemática informó que las anomalías de la sustancia blanca aparecen en el 41.3% de los casos de ARS 4). Las mutaciones de FOXC1 pueden inducir enfermedad de pequeños vasos cerebrales (CSVD), hiperintensidades de la sustancia blanca, espacios perivasculares agrandados, microhemorragias e infartos lacunares.
Ohkubo et al. (2025) confirmaron lesiones de la sustancia blanca periventricular, espacios perivasculares agrandados y dolicoectasia vertebrobasilar en la RM cerebral de un niño japonés de 2 años (mutación FOXC1: c.240del, p.Y81Ifs21). Su padre tenía antecedentes de infarto cerebral a los 18 años 4).
En una revisión de 95 casos con mutación de FOXC1, el 6.3% presentó síntomas neuropsiquiátricos (dificultades de aprendizaje, epilepsia, discapacidad intelectual, celos delirantes, etc.), y el 83.3% de los casos con mutaciones en el dominio forkhead mostraron síntomas neuropsiquiátricos 2).
Sí. Una revisión sistemática informó que las anomalías de la sustancia blanca aparecen en aproximadamente el 41% de los casos de ARS con mutaciones de FOXC1 4). Se ha sugerido que las mutaciones de FOXC1 están asociadas con enfermedad de pequeños vasos cerebrales y riesgo de accidente cerebrovascular, y el seguimiento neurológico a largo plazo es particularmente importante en ARS con mutación de FOXC1.
En los últimos años, se han reportado muchas mutaciones nuevas mediante secuenciación de próxima generación y secuenciación del genoma completo.
Wowra et al. (2024) identificaron una deleción grande (mutación nueva) que involucra parte del exón 1 de FOXC1 y todo el 3’UTR en tres hermanas polacas con ARS. Incluso dentro de la misma familia, los fenotipos variaron mucho, y inicialmente fueron diagnosticadas erróneamente con síndrome de Chandler 1).
Jiang et al. (2024) identificaron un reordenamiento genómico complejo en una familia china con ARS tipo 1, que incluye una deleción de 6.15 Mb del cromosoma 4q25 que contiene PITX2, una inversión de 45.71 Mb y una deleción de 14 pb. Una niña de 11 años presentó PIO de 43.5/44.0 mmHg 3).
Otros informes de mutaciones nuevas incluyen FOXC1 p.Phe136Leu (dominio forkhead) 2), FOXC1 p.S82R (mutación de novo) 5) y FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino et al. (2024) reportaron un caso de ARS tipo 3 en un hombre japonés de 77 años. Desarrolló delirios de celos a los 72 años y se confirmó leucoencefalopatía. En una revisión bibliográfica de 95 casos con mutaciones en FOXC1, se observaron síntomas neuropsiquiátricos en el 6.3% (6/95), y el 83.3% (5/6) de ellos tenían mutaciones en el dominio forkhead 2).
Este hallazgo sugiere que el dominio funcional de las mutaciones de FOXC1 puede estar involucrado en el desarrollo de síntomas neuropsiquiátricos, destacando la importancia del seguimiento a largo plazo desde una perspectiva de salud mental.
Se ha señalado que las mutaciones de FOXC1 pueden inducir CSVD y aumentar el riesgo de accidente cerebrovascular 4). La importancia de la prevención y la intervención temprana de las enfermedades neurovasculares en pacientes con ARS es un tema de investigación futuro.
Se está dilucidando la patogénesis de la “esclerización” corneal debida a mutaciones de FOXC1 1). Se espera que la comprensión de los mecanismos moleculares de la opacidad corneal conduzca al desarrollo de nuevas terapias que utilicen terapia génica, fármacos antifibróticos y biomateriales 1).