ARS tipo 1
Gene causador: PITX2 (4q25)
Anormalidades principais: Anormalidades do segmento anterior do olho, anormalidades dentárias, excesso de pele periumbilical/hérnia umbilical, anormalidades craniofaciais, anormalidades cardiovasculares

A síndrome de Axenfeld-Rieger (ARS) é um grupo de doenças congênitas que combinam anomalias do segmento anterior e anomalias sistêmicas. A causa fundamental é um distúrbio na migração e diferenciação das células da crista neural. No final da gestação, o processo de desaparecimento das células endoteliais indiferenciadas que cobrem a câmara anterior da íris e do ângulo é prejudicado, e seus remanescentes causam a formação de cordões ou inserção alta da íris.
Histórico: Em 1920, Axenfeld descreveu o anel de Schwalbe posterior (deslocamento anterior e espessamento da linha de Schwalbe) e processos da íris. Em 1934-1935, Rieger adicionou relatos de hipoplasia da íris, desvio pupilar e policoria. Atualmente é classificado em três estágios:
Tudo isso é coletivamente chamado de síndrome de Axenfeld-Rieger. 50-60% são acompanhados de glaucoma, é de herança autossômica dominante e geralmente bilateral. Frequentemente também está associada a catarata e ectopia lentis.
Epidemiologia: A prevalência foi estimada em cerca de 1/200.000, mas relatos recentes sugerem estimativas de 1/50.000 a 1/100.0002)4). Não há diferença entre sexos, e é frequentemente diagnosticado na infância ou primeira infância.
Classificação genética é a seguinte:
ARS tipo 1
Gene causador: PITX2 (4q25)
Anormalidades principais: Anormalidades do segmento anterior do olho, anormalidades dentárias, excesso de pele periumbilical/hérnia umbilical, anormalidades craniofaciais, anormalidades cardiovasculares
ARS tipo 2
Gene causador: 13q14 (não confirmado)
Anormalidades principais: Anormalidades do segmento anterior do olho, glaucoma. Anormalidades sistêmicas são menos frequentes que nos tipos 1 e 3
ARS tipo 3
Gene causador: FOXC1 (6p25)
Anormalidades principais: Anormalidades do segmento anterior do olho, glaucoma, perda auditiva neurossensorial, defeito do septo atrial, anormalidades renais, lesões da substância branca
Mutações em FOXC1 e PITX2 representam 40–70% dos casos de ARS5). No entanto, em 60% dos casos de ARS, o gene causador ainda não foi identificado4), indicando grande diversidade genética.
Em uma análise de grande registro de glaucoma de início na infância e em adultos jovens, a taxa de diagnóstico molecular foi de 56,5%11). Mutações em FOXC1 representaram 20,3%, PITX2 17,4% e PAX6 10,1%, e muitos casos não podem ser explicados por genes conhecidos11).
Eles são diferenciados pelo gene causador. O tipo 1 é causado por mutação em PITX2 (4q25) e acompanhado por anormalidades dentárias, umbilicais e ósseas faciais. O tipo 3 é causado por mutação em FOXC1 (6p25) e acompanhado por perda auditiva, defeitos cardíacos, anormalidades renais e neurológicas. O tipo 2 está localizado em 13q14, mas o gene causador não é confirmado, com anormalidades do segmento anterior e glaucoma como características principais. O diagnóstico pode ser confirmado por teste genético.
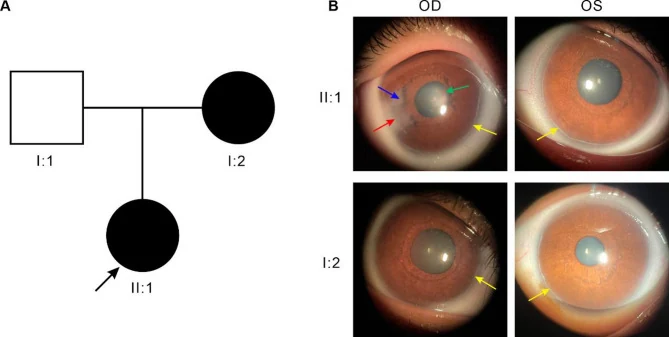
Principais itens dos achados oculares são mostrados abaixo.
| Achado ocular | Características |
|---|---|
| Anel embrionário posterior | Deslocamento anterior e espessamento da linha de Schwalbe |
| Processos da íris | Filiformes finos a largos em forma de faixa |
| Desvio pupilar | Desvio na direção oposta ao anel embrionário posterior |
| Pseudopolicoria | Aparência de perfuração no estroma da íris |
| Eversão uveal | Eversão do epitélio pigmentar da íris |
Anel embrionário posterior é um remanescente de células indiferenciadas na linha de Schwalbe, aparecendo linearmente ao longo do limbo, 0,5–2,0 mm centralmente ao limbo. Geralmente é parcial, não circunferencial. Se a linha de Schwalbe proeminente aderir à íris, é chamado de anomalia de Axenfeld; se acompanhado de atrofia do estroma da íris, é chamado de anomalia de Rieger.
A córnea geralmente é transparente com estrutura endotelial normal, mas o contato físico com tecido residual pode causar opacidade corneana secundária. A opacidade geralmente é limitada à periferia e geralmente não afeta diretamente a visão. No entanto, em mutações FOXC1, a opacidade e a neovascularização corneana são mais pronunciadas, e o grau de anormalidade corneana e a frequência de glaucoma são maiores em comparação com mutações PITX21).
Achados do ângulo incluem inserção alta da íris, remanescentes uveais em cordão e espessamento da linha de Schwalbe (anel embrionário posterior). Também foram relatados casos com microesferofacia ou subluxação do cristalino7).
Glaucoma ocorre em 50–60% dos casos. O aumento da pressão intraocular pode ocorrer desde a infância, mas a maioria se manifesta na infância ou idade adulta jovem. Alguns casos são diagnosticados após perda progressiva da visão, sendo importante não perder os achados do segmento anterior e as alterações glaucomatosas6).
No caso de Li et al. (2021) de um menino de 7 anos (ARS tipo 3, mutação de novo FOXC1), o diâmetro da córnea era 14 mm, comprimento axial 27,16/26,56 mm, relação C/D 0,9, PIO 33/20 mmHg. Ambos os olhos necessitaram de cirurgia antiglaucoma a partir do 36º dia de vida5).
Achados sistêmicos são os seguintes:
Cerca de 50-60% dos pacientes com ARS desenvolvem glaucoma. Geralmente surge na infância até o início da idade adulta, mas há casos com aumento da pressão intraocular desde a lactância. São necessárias medições regulares da pressão intraocular e avaliação do nervo óptico. Para detalhes, consulte a seção “Métodos de tratamento padrão”.
A ARS apresenta herança autossômica dominante com penetrância completa. No entanto, mesmo dentro da mesma família com a mesma mutação genética, há grande variabilidade clínica (expressividade variável)1).
Em um estudo de coorte de grande escala, mutações FOXC1 e PITX2 foram associadas a um amplo espectro de glaucoma desde a infância até a idade adulta9). Casos inicialmente diagnosticados clinicamente como glaucoma congênito primário (PCG) podem ser reclassificados por teste genético, e o teste genético contribui para o diagnóstico preciso do tipo quando os achados do segmento anterior em lactentes são sutis.
Em casos com microdeleção ao redor do gene PITX2, a sobreposição das deleções de NEUROG2, UGT8 e NDST4 pode causar atraso no desenvolvimento e deficiência intelectual 8)3).
Kawanami et al. (2023) relataram um menino japonês de 3 anos com microdeleção de 2,5 Mb em 4q25 (incluindo PITX2, NEUROG2 e ANK2). Ele apresentava hérnia umbilical, coloboma de íris e atraso no desenvolvimento, mas o ECG era normal apesar da deleção de ANK2. A haploinsuficiência de NEUROG2 foi considerada candidata a causa do atraso no desenvolvimento 8).
Por ser autossômica dominante, a probabilidade de herdar a mutação de um dos pais é de 50%. A penetrância é completa, mas o fenótipo varia, e a gravidade dos sintomas pode diferir significativamente mesmo com a mesma mutação 1). Exame genético e aconselhamento genético são recomendados.
O diagnóstico de ARS baseia-se em anomalias do ângulo da câmara anterior e da íris em ambos os olhos. A presença de embriotóxon posterior com aderência parcial da íris circundante é condição para o diagnóstico 13). Se o embriotóxon posterior não for visível na lâmpada de fenda, é necessária gonioscopia. Se houver anomalias sistêmicas, o paciente é encaminhado ao pediatra para avaliação completa como parte da síndrome ARS 13).
Note-se que 8-15% da população normal apresenta embriotóxon posterior leve, mas isso isoladamente não está associado a glaucoma ou outras condições. A história familiar também é importante no diagnóstico.
No sistema de classificação do glaucoma pediátrico de acordo com as Diretrizes de Tratamento do Glaucoma (5ª edição), a ARS é classificada como um exemplo típico de glaucoma associado a anomalias do desenvolvimento ocular congênito 13). O diagnóstico é feito quando as anomalias oculares presentes desde o nascimento preenchem os critérios diagnósticos de glaucoma pediátrico.
As principais doenças que devem ser diferenciadas da ARS são mostradas abaixo.
| Doença | Diferença da ARS |
|---|---|
| Síndrome ICE | Unilateral, adquirida, predominância feminina |
| Anomalia de Peters | Opacidade central da córnea, defeito da membrana de Descemet |
| Aniridia | Pannus corneano, hipoplasia foveal |
| Distrofia polimorfa posterior da córnea | Bilateral, familiar, sem diferença de sexo |
A síndrome ICE (como atrofia progressiva da íris, síndrome de Chandler) é importante para diferenciar da ARS, mas a principal diferença é que a ICE é unilateral e adquirida, enquanto a ARS é bilateral e congênita.
Atualmente não existe tratamento curativo para a ARS em si, e o manejo do glaucoma e a vigilância de complicações sistêmicas são o foco do tratamento. A abordagem terapêutica segue o glaucoma de desenvolvimento precoce (glaucoma congênito primário: PCG) 13).
O glaucoma ocorre em cerca de 50-60% dos casos de ARS. O tratamento medicamentoso segue as diretrizes gerais do glaucoma, mas é frequentemente ineficaz.
Inibidores da Produção de Humor Aquoso
Betabloqueadores: Uma das primeiras escolhas. Seguros e eficazes, mas frequentemente ineficazes em crianças.
Colírios inibidores da anidrase carbônica (CAI): como brinzolamida. Podem ser combinados com betabloqueadores.
Agonistas alfa-2 (brimonidina): Contraindicados em menores de 2 anos devido a efeitos neuropsiquiátricos (apneia, bradicardia, hipotensão, hipotonia, depressão do SNC) 13).
Medicamentos Facilitadores do Escoamento do Humor Aquoso
Análogos de prostaglandina: como latanoprost, travoprost. O efeito em crianças é mais fraco comparado aos adultos 13).
Exemplo: Em um menino de 7 anos, manejo de longo prazo com travoprost + brinzolamida 5). Em um homem de 77 anos, PIO de 35 mmHg mesmo com latanoprost/timolol + brinzolamida, indicando controle difícil 2).
Há relatos de que não há diferença na eficácia entre agonistas do receptor FP de prostanoides e betabloqueadores 13).
Em lactentes e crianças pequenas, a dose do colírio é relativamente grande em relação ao peso e área de superfície corporal, portanto deve-se usar a menor concentração possível do medicamento 13).
Se o controle da pressão intraocular não for alcançado com medicamentos, a cirurgia é realizada10)13).
As complicações pós-GDD incluem: câmara anterior rasa 13,6%, hipotonia 11,7%, efusão coroidal 8,3% e endoftalmite 1,7%14).
Chakraborty et al. (2022) relataram um caso de descolamento de retina associado à ARS (menino de 15 anos). Acompanhado de microfaquia e subluxação do cristalino, após vitrectomia a PIO aumentou para 41 mmHg e formou-se um estafiloma. Foi realizada fotocoagulação do corpo ciliar com diodo e, finalmente, a PIO atingiu 18 mmHg7).
A taxa de sucesso da cirurgia de ângulo é menor do que no glaucoma congênito primário 13). Na trabeculectomia com MMC, a taxa de sucesso em longo prazo de 2 anos é de aproximadamente 59%; nos dispositivos de drenagem para glaucoma, é relatada como 87% em 12 meses e 77% em 24 meses 14). Casos refratários podem necessitar de múltiplas cirurgias.
A causa fundamental da ARS é um defeito na migração e diferenciação das células da crista neural. O desenvolvimento prejudicado das células da crista neural na câmara anterior, ângulo do segmento anterior, ossos faciais, dentes, sistema cardiovascular e pele periumbilical resulta em malformações multiorgânicas.
No final da gestação, as células endoteliais indiferenciadas que revestem a câmara anterior normalmente desaparecem da íris e do ângulo. Na ARS, esse processo de desaparecimento é prejudicado, e as células endoteliais indiferenciadas persistem na íris, causando a formação de bandas. No ângulo, ocorre inserção alta da íris, cobrindo mecanicamente a malha trabecular.
Histologicamente, há uma camada única de células semelhantes a endotélio com uma membrana semelhante à membrana de Descemet que se estende anormalmente da superfície posterior da córnea através da câmara anterior, ângulo e superfície da íris. A membrana está presente nos quadrantes com eversão uveal e desvio pupilar, enquanto atrofia da íris é observada nos quadrantes opostos.
FOXC1 e PITX2 são ambos fatores de transcrição, que se ligam a sequências específicas de DNA e regulam a expressão de genes a jusante. Ambos atuam sinergicamente no desenvolvimento do segmento anterior do olho e regulam genes-alvo comuns a jusante 3). O domínio forkhead do FOXC1 (domínio de ligação ao DNA de 110 aminoácidos) é o mais importante funcionalmente 2), e mutações nesse domínio são sugeridas como mais fortemente associadas a sintomas neuropsiquiátricos.
Dois mecanismos de elevação da pressão intraocular foram identificados:
O grau de defeito da íris e a quantidade de processos irianos no ângulo não se correlacionam necessariamente com a gravidade do glaucoma. No entanto, a adesão iriana grave no ângulo aumenta o risco de glaucoma.
A mutação FOXC1 promove mais o glaucoma congênito do que outras mutações 1), e anormalidades morfológicas do corpo ciliar e do ângulo de drenagem podem contribuir para o aumento da PIO 1).
Em camundongos com mutação FOXC1, observou-se diminuição das fibras de colágeno do estroma corneano, anormalidades estruturais e danos às células do estroma 1). Além disso, FOXC1 funciona como um fator supressor da neovascularização corneana (através do controle da biodisponibilidade do VEGF) 1), e a perda dessa supressão devido à mutação FOXC1 leva à neovascularização corneana.
FOXC1, como fator de transcrição da família FOX, também desempenha um papel importante no desenvolvimento cerebral 4).
Em uma revisão sistemática, foi relatado que anormalidades da substância branca aparecem em 41,3% dos casos de ARS 4). A mutação FOXC1 pode induzir doença de pequenos vasos cerebrais, hiperintensidade da substância branca, alargamento dos espaços perivasculares, micro-hemorragias e infartos lacunares.
Ohkubo et al. (2025) confirmaram lesões da substância branca periventricular, alargamento dos espaços perivasculares e tortuosidade e ectasia da artéria vertebrobasilar em ressonância magnética cerebral de um menino japonês de 2 anos (mutação FOXC1: c.240del, p.Y81Ifs21). O pai tinha histórico de infarto cerebral aos 18 anos 4).
Em uma revisão de 95 casos de mutação FOXC1, sintomas neuropsiquiátricos (dificuldades de aprendizagem, epilepsia, deficiência intelectual, delírio de ciúmes, etc.) foram encontrados em 6,3%, e sintomas neuropsiquiátricos apareceram em 83,3% dos casos com mutações no domínio forkhead 2).
Sim. Há uma revisão sistemática que relata que anormalidades da substância branca aparecem em cerca de 41% dos casos de ARS com mutação FOXC1 4). A mutação FOXC1 pode estar associada a doença de pequenos vasos cerebrais e risco de acidente vascular cerebral, e especialmente na ARS tipo FOXC1, o acompanhamento neurológico de longo prazo é importante.
Nos últimos anos, muitas novas mutações foram relatadas por meio de sequenciamento de próxima geração e sequenciamento do genoma completo.
Wowra et al. (2024) identificaram uma grande deleção (nova mutação) envolvendo parte do éxon 1 e toda a região 3’UTR do FOXC1 em três irmãs polonesas com ARS. O fenótipo variou amplamente mesmo dentro da mesma família, e elas foram inicialmente diagnosticadas erroneamente com síndrome de Chandler 1).
Jiang et al. (2024) identificaram um rearranjo genômico complexo consistindo em uma deleção de 6,15 Mb no cromossomo 4q25 envolvendo PITX2, uma inversão de 45,71 Mb e uma deleção de 14 bp em uma família chinesa com ARS tipo 1. Uma menina de 11 anos apresentou PIO de 43,5/44,0 mmHg 3).
Outros relatos de novas mutações incluem FOXC1 p.Phe136Leu (domínio forkhead) 2), FOXC1 p.S82R (mutação de novo) 5), e FOXC1 c.240del, p.Y81Ifs21 4).
Yoshino et al. (2024) relataram um caso de um homem japonês de 77 anos com ARS tipo 3. Delírios de ciúme apareceram aos 72 anos, e leucoencefalopatia foi confirmada. Em uma revisão de literatura de 95 casos de mutação FOXC1, sintomas neuropsiquiátricos foram encontrados em 6,3% (6/95), e 83,3% (5/6) deles tinham mutações no domínio forkhead 2).
Essa descoberta sugere que o domínio funcional da mutação FOXC1 pode estar envolvido na manifestação de sintomas neuropsiquiátricos, indicando a importância do acompanhamento de longo prazo sob a perspectiva da saúde mental.
Mutações FOXC1 foram apontadas como indutoras de CSVD e aumento do risco de acidente vascular cerebral 4). A prevenção e intervenção precoce de doenças neurovasculares em pacientes com ARS são tópicos de pesquisa futura.
A compreensão da patogênese da “esclerotização” da córnea causada por mutações FOXC1 está avançando 1). Espera-se que o entendimento dos mecanismos moleculares da opacidade da córnea leve ao desenvolvimento de novos tratamentos usando terapia gênica, drogas antifibróticas e biomateriais 1).