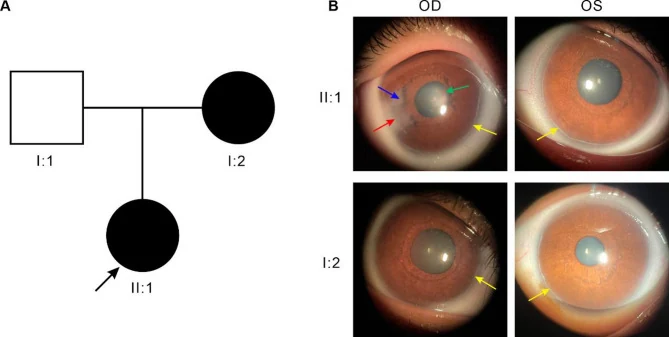
Gong X, Lei X, Li Z, et al. Identification and functional study of a novel FOXC1 missense mutation in a Chinese family with Axenfeld-Rieger syndrome. Sci Rep. 2025;15(1):19957. Fig. 1B. PMID: 40481210; PMCID: PMC12144153; DOI: 10.1038/s41598-025-04872-x. License: CC BY.
Kawanami et al.(2023)は、4q25に2.5Mbの微小欠失(PITX2・NEUROG2・ANK2を含む)を有する3歳日本人男児を報告した。臍帯ヘルニア・虹彩コロボーマ・発達遅延を呈したが、ANK2欠失にもかかわらず心電図は正常であった。NEUROG2のハプロ不全が発達遅滞の原因候補とされた8)。
Wowra B, Wysocka-Kosmulska M, Stanienda-Sokół K, et al. Diagnostic Challenges of Axenfeld-Rieger Syndrome and a Novel FOXC1 Gene Mutation in a Polish Family. J Clin Med. 2024;13(19):5761. doi:10.3390/jcm13195761.
Yoshino Y, Iga JI, Ueno SI.. A Novel Mutation of FOXC1 (P136L) in an Axenfeld-Rieger Syndrome Patient With a Systematized Delusion of Jealousy: A Case Report and Literature Review. Mol Genet Genomic Med. 2024;12(11):e70008. doi:10.1002/mgg3.70008. PMID:39520097; PMCID:PMC11549375.
Jiang Z, Zhang Y, Wang L, Yang H, Yu L. Complex genomic rearrangement with deletion of PITX2 in a Chinese family with Axenfeld-Rieger syndrome: A case report and literature review. Molecular vision. 2024;30:466-476. PMID:39959165; PMCID:PMC11829790.
Ohkubo Y, Hayashi T, Ida K, Nakano E, Hamada A, Higashida T, et al. A case of Axenfeld-Rieger syndrome with neuroradiological abnormalities. Radiology case reports. 2025;20(10):5078-5080. doi:10.1016/j.radcr.2025.06.072. PMID:40741157; PMCID:PMC12310057.
Li K, Tang M, Xu M, Yu Y. A novel missense mutation of FOXC1 in an Axenfeld-Rieger syndrome patient with a congenital atrial septal defect and sublingual cyst: a case report and literature review. BMC Med Genomics. 2021;14:255. doi:10.1186/s12920-021-01103-w.
Khan TA, Zahid MA, Akram A, Rauf A. Progressive Vision Loss in a Patient With Axenfeld-Rieger Syndrome. Cureus. 2022;14(5):e25128. doi:10.7759/cureus.25128.
Chakraborty D, Basak SK, Saha A, Mondal S, Mitra A. A case of Axenfeld-Rieger syndrome with retinal detachment. Indian journal of ophthalmology. 2022;70(7):2650-2652. doi:10.4103/ijo.IJO_2737_21. PMID:35791188; PMCID:PMC9426138.
Kawanami Y, Horinouchi T, Morisada N, Kato T, Nozu K. 4q25 Microdeletion with Axenfeld-Rieger Syndrome and Developmental Delay. Case reports in genetics. 2023;2023:4592114. doi:10.1155/2023/4592114. PMID:36816813; PMCID:PMC9935865.
Souzeau E, Siggs OM, Zhou T, Galanopoulos A, Hodson T, Taranath D, et al. Glaucoma spectrum and age-related prevalence of individuals with FOXC1 and PITX2 variants. European journal of human genetics : EJHG. 2017;25(7):839-847. doi:10.1038/ejhg.2017.59. PMID:28513611; PMCID:PMC5520071.
Knight LSW, Ridge B, Staffieri SE, Ruddle JB, Taranath DA, Craig JE, Mackey DA, Burdon KP. Childhood and early onset glaucoma classification and genetic profile in a large Australasian disease registry. Ophthalmology. 2021;128(11):1549-1559. doi:10.1016/j.ophtha.2021.04.016.
Mirzayans F, Gould DB, Héon E, Billingsley GD, Cheung JC, Mears AJ, et al. Axenfeld-Rieger syndrome resulting from mutation of the FKHL7 gene on chromosome 6p25. European journal of human genetics : EJHG. 2000;8(1):71-4. doi:10.1038/sj.ejhg.5200354. PMID:10713890.
Stallworth JY, O’Brien KS, Han Y, Oatts JT. Efficacy of Ahmed and Baerveldt glaucoma drainage device implantation in the pediatric population: A systematic review and meta-analysis. Surv Ophthalmol. 2023;68(4):578-590. doi:10.1016/j.survophthal.2023.01.010. PMID:36740196; PMCID:PMC10293048.