先天性合併症(出生時から)

無虹彩症(Aniridia)
1. 無虹彩症とは
Section titled “1. 無虹彩症とは”無虹彩症(aniridia)は、先天的な素因によって虹彩が完全または不完全に欠損した状態である。「無虹彩」と呼ばれるが、隅角の最周辺部に虹彩根部が残存することが多い。
2017年に厚生労働省の難病法に基づく指定難病として認定された1)。指定難病として診断され、重症度分類がⅢ度以上と判断された患者は医療費助成の対象となり、所得に応じた自己負担上限額が設定される2)。
| 項目 | 内容 |
|---|---|
| 有病率 | 6.4万〜9.6万人に1人1) |
| 性差 | なし1) |
| 両眼性 | 60〜90%1) |
| 遺伝形式(家族性) | 全体の約2/3(常染色体優性遺伝) |
| 孤発性 | 全体の約1/3 |
| Wilms腫瘍合併(孤発例) | 約30%(WAGR症候群)3) |
スウェーデン・ノルウェーの疫学研究では有病率は約90,000人に1人と報告されている3)。PAX6遺伝子変異を有する43例の詳細な眼科的評価では、虹彩形成異常の程度は変異の種類によって異なることが示されている3)。
Q
無虹彩症は遺伝しますか?
A
全体の約2/3は常染色体優性遺伝であり、罹患した親から子へ50%の確率で遺伝する可能性がある。残り1/3は孤発性で家族歴がない。孤発例ではWilms腫瘍(腎腫瘍)を合併するWAGR症候群のリスクがあるため、PAX6遺伝子とWT1遺伝子の遺伝学的検査が推奨される。
2. 主な症状と臨床所見
Section titled “2. 主な症状と臨床所見”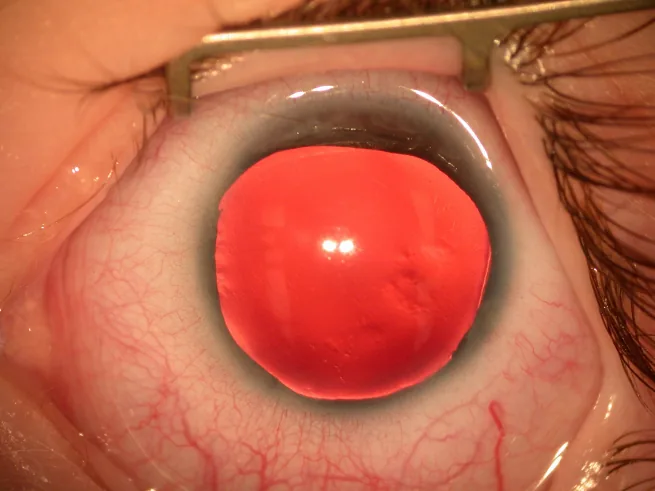
Law SK, et al. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol Vis. 2011. Figure 2. PMCID: PMC3102021. License: CC BY.
虹彩が欠損または不完全なため瞳孔が機能せず、眼球に入る光量を調節できない。そのため強い羞明を訴える。また黄斑低形成による固視不良が、生後早期からみられる水平眼振の主訴となることが多い。
後天性合併症(成長に伴い発症)
眼合併症の頻度まとめ
Section titled “眼合併症の頻度まとめ”| 合併症 | 頻度・時期 | 視機能への影響 |
|---|---|---|
| 黄斑低形成 | ほぼ全例(先天性) | 最大の視力制限因子。有効な治療法なし |
| 白内障 | 約80%(後天性)1) | 視力低下・羞明の増悪 |
| 緑内障 | 50〜75%(後天性)1) | 進行すれば不可逆的視野障害 |
| 角膜輪部疲弊症 | 成長後に発症・進行3) | 角膜実質混濁→高度視力低下 |
| 眼球振盪症 | 先天性(ほぼ全例) | 固視不良 |
| 斜視 | 先天性〜乳幼児期 | 弱視のリスク |
PAX6遺伝子は眼組織のほか、中枢神経・膵臓Langerhans島・嗅上皮にも発現している。これらの組織の低形成により多様な眼外合併症を伴うことがある1)。
- 脳梁欠損、てんかん、高次脳機能障害
- 無嗅覚症
- グルコース不耐性
- WAGR症候群(孤発例の約30%):Wilms腫瘍・無虹彩・泌尿生殖器異常・精神遅滞3)
Q
無虹彩症でどのくらい見えますか?
A
視力予後はおおむね不良で0.1程度となることが多い。ただし黄斑低形成の程度や合併症の有無によって0.1〜0.7まで個人差がある。黄斑低形成は現時点で有効な治療法がなく最大の視力制限因子となる。適切な屈折矯正とロービジョンケアにより日常生活の質を改善できる。
3. 原因とリスク要因
Section titled “3. 原因とリスク要因”病因:PAX6遺伝子のハプロ不全
Section titled “病因:PAX6遺伝子のハプロ不全”無虹彩症の原因は第11染色体短腕(11p13)に位置するPAX6遺伝子の片アリルの機能喪失(ハプロ不全)による。機能的遺伝子量が半減することで生じる。両アリルが異常な場合は胎生致死となると考えられている1)。
PAX6は胎生期の器官分化を司る転写因子のマスター・コントロール遺伝子であり、さまざまな転写因子を統括する。PAX6の異常によって眼球全体にわたる種々の先天異常(無虹彩・Peters異常・黄斑低形成など)が起こる。
遺伝子変異の型はナンセンス・フレームシフトなどpremature truncated codon(PTC)型の変異が多く、ミスセンス変異も報告されている1)。isolated aniridiのシークエンシング解析ではPAX6変異が約85%に検出される2)。
WAGR症候群(孤発例の注意点)
Section titled “WAGR症候群(孤発例の注意点)”PAX6遺伝子はがん抑制遺伝子であるWT1遺伝子と11p13染色体上で隣接している。孤発例では隣接遺伝子欠失により、Wilms腫瘍・無虹彩・泌尿生殖器異常・精神遅滞(mental retardation)からなるWAGR症候群を呈することがある3)。孤発例の約30%でWilms腫瘍が5歳までに早期両側発症する。
遺伝カウンセリングの要点
Section titled “遺伝カウンセリングの要点”- PAX6変異陽性でWT1欠失なし→WAGR症候群の可能性はないと推定できる2)
- 遺伝学的検査はDNAシークエンシング+MLPA/CMAによるゲノム構造異常検出の組み合わせを行う2)
- 孤発例でWAGR症候群が疑われる場合は遺伝学的検査が推奨される2)
Q
無虹彩症の遺伝子検査は受けるべきですか?
A
PAX6遺伝子検査はDefinite診断の確定に必要であり、特に孤発例ではWilms腫瘍リスク評価のためPAX6とWT1の遺伝学的検査が推奨される。検査はDNAシークエンシングとMLPA/CMAを組み合わせ、適切な遺伝カウンセリングのもとで実施することが重要である。
4. 診断と検査方法
Section titled “4. 診断と検査方法”診断基準(厚労省指定難病 2020)
Section titled “診断基準(厚労省指定難病 2020)”無虹彩症の診断基準および重症度分類1)によるカテゴリー分類を以下に示す。
| 診断カテゴリー | 診断基準の組み合わせ |
|---|---|
| Definite | Aのいずれか+B1+E を満たし、Cを除外したもの |
| Probable (1) | Aのいずれか+B1+F を満たし、Cを除外したもの |
| Probable (2) | Aのいずれか+B1+B2 を満たし、Cを除外したもの |
| Probable (3) | Aのいずれか+B1+B3 を満たし、Cを除外したもの |
| Possible | Aのいずれか+B1 を満たし、Cを完全には除外できないもの |
A. 症状
- 両眼性の視力障害(黄斑低形成・白内障・緑内障・角膜輪部疲弊症による視力低下)
- 羞明(虹彩欠損の程度により)
B. 検査所見
- 細隙灯顕微鏡検査で部分的虹彩萎縮から完全虹彩欠損までさまざまな程度の虹彩の形成異常(60〜90%が両眼性)
- 眼底検査・OCT検査で黄斑低形成(中心窩陥凹・黄斑色素・中心窩無血管領域が不明瞭)
- 細隙灯顕微鏡検査で角膜輪部疲弊症や角膜混濁等の角膜病変
- 細隙灯顕微鏡検査で白内障(約80%に合併)
- 超音波検査・MRI・CTで小眼球
- 眼球振盪症
- 眼圧検査等で緑内障(50〜75%に合併)
C. 鑑別診断(除外すべき疾患)
- ヘルペスウイルス科の既感染による虹彩萎縮
- 外傷後または眼内手術後の虹彩欠損
- 眼杯裂閉鎖不全に伴う虹彩コロボーマ
- Rieger異常
- 虹彩角膜内皮(ICE)症候群
D. PAX6遺伝子変異に伴う眼外合併症(脳梁欠損・てんかん等)
E. PAX6遺伝子の病的遺伝子変異もしくは11p13領域の欠失(遺伝学的検査)
F. 家族内発症(常染色体優性遺伝が2/3)
| 検査 | 目的・内容 |
|---|---|
| 細隙灯顕微鏡検査 | 虹彩形成異常の程度評価(診断の基本) |
| 眼底検査・OCT | 黄斑低形成(中心窩陥凹消失・黄斑色素不明瞭)の評価 |
| 隅角鏡検査 | 隅角形成不全・残存虹彩根部と線維柱帯の癒着評価 |
| 眼圧測定(定期) | 緑内障スクリーニング。青年期から定期的に実施 |
| 腹部超音波検査 | Wilms腫瘍スクリーニング(孤発例、数か月ごと、特に5歳まで) |
| 遺伝子検査 | PAX6遺伝子変異または11p13領域欠失の同定(Definite診断に必要) |
小児では全身麻酔下検査が必要になる場合がある。
Q
無虹彩症の診断はどのように行いますか?
5. 標準的な治療法
Section titled “5. 標準的な治療法”治療の全体方針
Section titled “治療の全体方針”虹彩形成異常・黄斑低形成・小眼球・眼球振盪症は現時点で介入不可であり、経過観察が基本となる。治療対象は角膜症・白内障・緑内障・羞明・ロービジョンである2)。
治療方針の概要
Section titled “治療方針の概要”無虹彩症では、角膜、白内障、緑内障、ロービジョン、羞明をそれぞれ分けて管理する2)。
| 治療領域 | 診療方針 |
|---|---|
| 角膜実質混濁 | 角膜移植は視機能改善が限定的で、適応を慎重に判断する |
| 角膜上皮幹細胞疲弊症 | 眼表面再建術を検討する |
| 白内障 | 混濁と羞明の程度をみて手術を検討する |
| 高眼圧・緑内障 | 視機能温存のため積極的に治療する |
| ロービジョンケア | 早期から導入する |
| 羞明 | 遮光眼鏡やコンタクトレンズなどで対策する |
角膜症の治療
Section titled “角膜症の治療”角膜実質混濁:角膜移植によって得られる視機能の改善は、無虹彩症の合併症により限定的である2)。長期的には緑内障悪化・経年的な移植片機能不全により視力予後が不良となる場合が多い。角膜混濁に対する全層角膜移植は視力改善に結びつかないことが多く、拒絶反応率が高いことに注意を要する。重症例では益と害のバランスを十分に検討したうえで施行を判断する。
角膜上皮幹細胞疲弊症(LSCD):手術加療を検討する2)。具体的には他家輪部移植(KLAL)または培養口腔粘膜上皮移植(COMET)によりある程度の眼表面再建が期待できる3)。角膜実質混濁を合併する場合には角膜移植の併用が視力向上に有用なことが多い2)。
白内障の治療
Section titled “白内障の治療”20歳までに50〜85%で白内障が発症し、混濁と羞明の強度により白内障手術を計画する2)。
- 水晶体嚢・Zinn小帯の脆弱性に伴う手術難度が高い
- 術後の緑内障悪化・anterior fibrosis syndrome・水疱性角膜症のリスクに注意する2)
- 眼内レンズ(IOL)挿入は慎重な適応となる3)
- 白内障手術と同時の人工虹彩挿入は緑内障を誘発する可能性があるため推奨されない
手術に伴うリスクについて十分な説明を行ったうえで実施する。
緑内障の治療
Section titled “緑内障の治療”緑内障は視機能予後に直結するため、積極的に治療する2)。段階的な以下のアプローチをとる。
- 薬物療法:副作用に留意し、小児への全身影響を考慮して点眼・内服による眼圧下降を行う
- 流出路再建術:隅角切開術・線維柱帯切開術(薬物療法が無効な場合に検討)
- 濾過手術:線維柱帯切除術
- 緑内障インプラント手術:ロングチューブ手術(施設認定が必要)
- 毛様体凝固術:他治療が奏功しない場合の最終手段
薬物治療への抵抗が多く、チューブシャント手術が良い選択肢となる可能性がある4)。緑内障は視野障害が不可逆的であるため、早期眼圧管理が視機能保持の鍵となる。
Q
無虹彩症の緑内障はどのように治療しますか?
A
まず点眼・内服による薬物療法を行うが、多くは薬物に抵抗する。効果不十分な場合は流出路再建術(隅角切開術・線維柱帯切開術)を検討し、さらに線維柱帯切除術やロングチューブ手術(緑内障インプラント手術)へ進む。ロングチューブ手術は施設認定が必要である。毛様体凝固術は他治療が奏功しない場合の最終手段となる。定期的な眼圧モニタリングが不可欠である。
ロービジョンケアと羞明対策
Section titled “ロービジョンケアと羞明対策”ロービジョンケアおよび羞明対策は、視機能と生活の質を保つために早期から導入する2)。
- 屈折矯正:屈折異常を眼鏡で補正し、可能な限り視覚発達を促す(基本)
- 遮光眼鏡:羞明軽減に有効。強い羞明がある場合に処方する
- 人工虹彩付きSCL:羞明の改善と外観改善の両面で有用
- 拡大鏡・弱視眼鏡・拡大読書器などの視覚補助具を活用する
6. 病態生理学・詳細な発症機序
Section titled “6. 病態生理学・詳細な発症機序”PAX6遺伝子と眼形成
Section titled “PAX6遺伝子と眼形成”PAX6遺伝子は胎生期の器官分化を司る転写因子をコードするマスター・コントロール遺伝子である。初期眼球から発現し、さまざまな転写因子を統括する。PAX6の片アリル機能喪失(ハプロ不全)によって眼球全体にわたる先天異常(無虹彩・Peters異常・黄斑低形成など)が起こる。
PAX6変異はナンセンス・フレームシフトなどPTC型が多く、ミスセンス変異も報告されている1)。遺伝型と表現型の相関(genotype-phenotype correlation)についての研究では、変異の種類によって眼科的所見の重症度が異なることが示されている3)。
PAX6は眼以外にも中枢神経・膵臓Langerhans島・嗅上皮に発現しており、各組織の低形成による眼外合併症(脳梁欠損・てんかん・無嗅覚症・グルコース不耐性)が出現しうる1)。
緑内障の発症機序
Section titled “緑内障の発症機序”無虹彩症に伴う緑内障の発症機序には2つの経路が考えられている。
- 開放隅角型病態:線維柱帯における房水流出抵抗の増大
- 閉塞隅角型病態:最周辺部に残存した虹彩根部が線維柱帯と癒着し、一種の閉塞隅角緑内障の病態をきたす
乳児期に緑内障を呈することはまれで、成長に伴い青年期に進行性に発症する。隅角の形成異常によって開放状態で起きる場合と、閉塞隅角によって緑内障を呈する場合がある。
角膜輪部疲弊症(LSCD)の病態
Section titled “角膜輪部疲弊症(LSCD)の病態”病理学的には角膜上皮幹細胞の機能異常がみられ、上皮とBowman膜に異常をきたし、血管豊富なパンスが形成される。palisades of Vogtの形成不全から結膜組織の侵入・角化へと進行する1)。
無虹彩症の角膜は健常者と比べて厚い。幼少時には角膜が正常であることが多いが、成長に伴い角膜実質混濁・LSCDを合併して視力低下の原因となる。14年間の単施設研究(738眼)ではLSCDの原因疾患のなかで無虹彩症が30.9%と最多であった6)。
- 視力予後はおおむね不良で0.1程度となることが多い
- 黄斑低形成は有効な治療法がなく最大の視力制限因子となる
- 緑内障による視野障害は不可逆的であり、早期眼圧管理が重要
- 孤発例ではWilms腫瘍の5歳までの早期発症に注意し、定期的な腹部超音波検査を継続する
長期予後に関する研究では、視力予後はおおむね不良であるが、合併症の種類と重症度によって個人差があることが報告されている5)。
7. 最新の研究と今後の展望
Section titled “7. 最新の研究と今後の展望”遺伝学的検査の進歩
Section titled “遺伝学的検査の進歩”次世代シークエンシング(NGS)の普及により、isolated aniridiに対するPAX6変異の検出率は約85%となっている2)。染色体マイクロアレイ(CMA)は11p13の微細欠失検出において従来の染色体検査より高感度であり、WAGR症候群の診断精度向上に貢献している2)。
培養口腔粘膜上皮移植(COMET)の長期成績の蓄積が進んでいる2)。Boston type I keratoprosthesisについては短期的(17〜28.7か月)に65〜93%で視力改善が得られるが、4.5年の時点では43.5%まで低下するという報告がある2)。
人工虹彩デバイスと遺伝子治療の展望
Section titled “人工虹彩デバイスと遺伝子治療の展望”HumanOptics CustomFlex ArtificialIrisは個別注文のシリコン製人工虹彩デバイスであり、羞明軽減と外観改善に有用とされているが、2024年現在、日本では未承認である。PAX6ハプロ不全を標的とした分子標的治療は現時点で研究段階にあり、臨床応用には至っていない3)。
8. 参考文献
Section titled “8. 参考文献”- 大家義則, 川崎諭, 西田希, 木下茂, 外園千恵, 大橋裕一, 他. 無虹彩症の診断基準および重症度分類. 日眼会誌. 2020;124:83-88.
- 厚生労働科学研究費補助金難治性疾患政策研究事業「角膜難病の標準的診断法および治療法の確立を目指した調査研究」研究班. 無虹彩症の診療ガイドライン. 日眼会誌. 2021;125:38-73.
- Hingorani M, Hanson I, van Heyningen V. Aniridia. Eur J Hum Genet. 2012 Oct;20(10):1011-1017. doi:10.1038/ejhg.2012.100. PMID:22692063; PMCID:PMC3449076.
- American Academy of Ophthalmology. Diagnosis and Management of Aniridia. EyeNet Magazine. 2014. https://www.aao.org/eyenet/article/diagnosis-management-of-aniridia
- Japanese Ophthalmological Society. Clinical practice guideline for aniridia. Jpn J Ophthalmol. 2026. doi:10.1007/s10384-025-01296-y. https://link.springer.com/article/10.1007/s10384-025-01296-y
- Hu JCW, Weissbart SB. Limbal stem cell deficiency and severe ocular surface disease: a review. Ann Eye Sci. 2023;8:35.