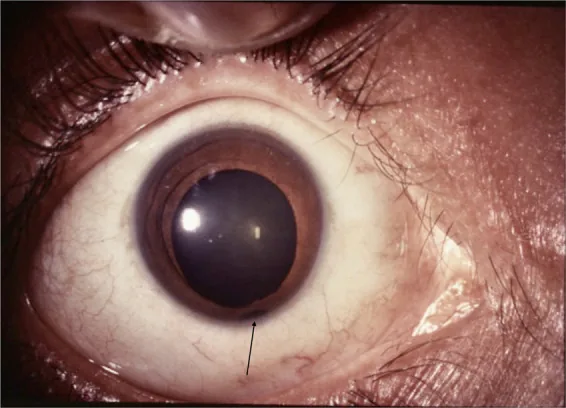
Scemla B, Duroi Q, Duraffour P, Souedan V, Brézin AP. Transscleral filtration revealing a chorioretinal coloboma. American journal of ophthalmology case reports. 2021;21:101003. doi:10.1016/j.ajoc.2020.101003. PMID:33385097; PMCID:PMC7771107.
Rajshri H, Nagesha CK, Arthi M. Combined choroidal vitiligo and retinochoroidal coloboma. BMJ case reports. 2023;16(4). doi:10.1136/bcr-2022-253854. PMID:37105595; PMCID:PMC10151958.
Ratra D, Mohan S, Nadig R, Kashyap H. The deceptive coloboma. Indian journal of ophthalmology. 2023;71(6):2632. doi:10.4103/IJO.IJO_250_23. PMID:37322728; PMCID:PMC10418011.
Castilla-Martinez G, Fernandez-Martinez C, Pardo-Lopez S, Toledano-Martos R, Hernandez-Artola F. FLACS (Femtosecond-Laser-Assisted Cataract Surgery) combined with pupiloplasty in iris-lens coloboma. American journal of ophthalmology case reports. 2024;34:102039. doi:10.1016/j.ajoc.2024.102039. PMID:38680527; PMCID:PMC11047286.
Hu X, Lin W, Luo Z, Zhong Y, Xiao X, Tang R. Frameshift Mutation in PAX2 Related to Focal Segmental Glomerular Sclerosis: A Case Report and Literature Review. Mol Genet Genomic Med. 2024;12:e70006. doi:10.1002/mgg3.70006. PMID:39235128; PMCID:PMC11375732.
Cortes-Gonzalez V, Rodriguez-Morales M, Ataliotis P, et al. Homozygosity for a hypomorphic mutation in frizzled class receptor 5 causes syndromic ocular coloboma with microcornea in humans. Hum Genet. 2024;143:1509-1521. doi:10.1007/s00439-024-02712-y. PMID:39503780; PMCID:PMC11576812.
Jain KS, Upadhyaya A, Raval VR. Fibrin-glue-assisted retinopexy for coloboma-associated retinal detachment. Indian journal of ophthalmology. 2024;72(12):1840. doi:10.4103/IJO.IJO_972_24. PMID:39620692; PMCID:PMC11727969.
Youfeng Zhou, Ke Xu, Weiyue Gu, Yan Huang. Microcornea, iris and choroidal coloboma, and global developmental delay caused by TENM3 pathogenic variants in a Chinese patient. Molec Gen & Gen Med. 2022;10(6). doi:10.1002/mgg3.1948.
Emran Esmaeilzadeh, Zhila Ghaderi, Arman Moradi, Hamid Reza Khorram Khorshid. Identification of causative gene mutation in an Iranian family with coloboma and nephropathy using whole exome sequencing. CEN Case Rep. 2022;11(4):404-407. doi:10.1007/s13730-022-00692-4.