عنبیه
مردمک کلیدیشکل: بهطور معمول نقص در سمت پایین-داخلی قرار دارد و مردمک به شکل کلید تغییر شکل میدهد.
نزدیک به سمت گیجگاهی-پایینی: ممکن است در موقعیتهای غیرمعمول نیز رخ دهد.

کولوبوما (نقص چشم) واژهای است که از کلمه یونانی به معنای «نقص» گرفته شده و یک بیماری مادرزادی است که در اثر بسته نشدن شکاف جنینی، در قسمتهای مختلف چشم ایجاد نقص بافتی میکند. این نقص میتواند در پلک، عنبیه، عدسی، جسم مژگانی، مشیمیه، شبکیه و عصب بینایی رخ دهد. نقص معمولاً در قسمت پایین-بینی قرار دارد و اغلب با میکروفتالمی (کوچکی چشم) همراه است.
شیوع این بیماری 0.5 تا 2.2 مورد در هر 10,000 تولد گزارش شده است. در ایالات متحده، حدود 2.6 مورد در هر 10,000 تولد 4) و در اروپا 4 تا 19 مورد در هر 100,000 تولد گزارش شده است 6). این بیماری حدود 11% از موارد نابینایی کودکان را تشکیل میدهد و میزان تشخیص ژنتیکی کمتر از 30% است 6). شیوع کلوبوم پلک 0.2 تا 0.8 مورد در هر 10,000 تولد است. نسبت آن در ناهنجاریهای مادرزادی چشم 0.07% و در کودکان مبتلا به اختلال بینایی 3.2 تا 11.2% گزارش شده است.
کولوبوما (نقص چشم) دو نوع دارد: نقص معمولی و نقص غیرمعمولی. نقص معمولی ناشی از بسته نشدن شکاف جنینی است و در سمت پایین-بینی قرار دارد، در حالی که نقص غیرمعمولی در سایر نواحی رخ میدهد و مکانیسم رشدی متفاوتی برای آن فرض میشود.
کد ICD-10 برای این بیماری Q10.3 (پلک)، Q13.0 (عنبیه)، Q12.2 (عدسی)، H47.319 (عصب بینایی) و Q14.8 (مشیمیه و شبکیه) است.
هم به صورت پراکنده و هم ارثی دیده میشود. الگوهای وراثتی متنوعی از جمله اتوزومال غالب، اتوزومال مغلوب و وابسته به X گزارش شده است. ژنهای عامل متعددی مانند PAX2، CHD7 و FZD5 شناسایی شدهاند، اما میزان تشخیص ژنتیکی کمتر از 30% است6). در صورت وجود سابقه خانوادگی، مشاوره ژنتیک توصیه میشود.
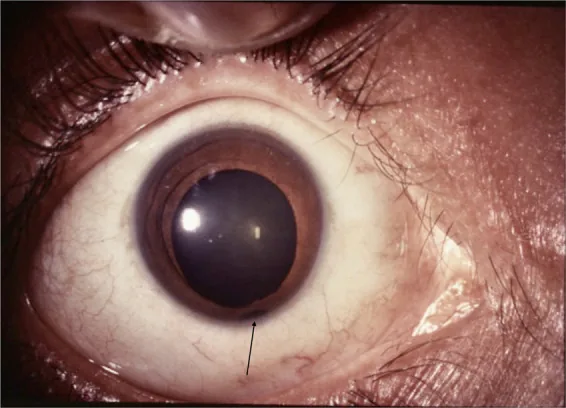
بینایی بسته به محل و وسعت نقص، از عدم درک نور تا طبیعی بودن، بسیار متفاوت است.
کولوبوما در هر بخش از چشم یافتههای مشخصی را نشان میدهد.
عنبیه
مردمک کلیدیشکل: بهطور معمول نقص در سمت پایین-داخلی قرار دارد و مردمک به شکل کلید تغییر شکل میدهد.
نزدیک به سمت گیجگاهی-پایینی: ممکن است در موقعیتهای غیرمعمول نیز رخ دهد.
مشیمیه و شبکیه
ضایعات زرد-سفید: نقصهای گرد تا بادبزنی با مرز مشخص که صلبیه از طریق آن قابل مشاهده است.
خطر جداشدگی شبکیه: میزان بروز 23 تا 40%7). پیگیری منظم ضروری است.
عصب بینایی و عدسی
بزرگشدگی حفره عصب بینایی: از یک طرفه تا دوطرفه، با درجات مختلف.
صاف شدن استوای عدسی: در نتیجه نقص زونولهای دارچینی ایجاد میشود. در حالت گشاد شدن مردمک مشاهده میگردد.
پلک
نقص در قسمت داخلی پلک فوقانی: نقص بافتی تماملایه.
ناهنجاریهای سیستمیک همراه: اگرچه موارد مجزا نیز وجود دارد، اما ممکن است با ناهنجاریهای سیستمیک همراه باشد.
کولوبومای جسم مژگانی به ندرت به تنهایی رخ میدهد و اغلب به صورت پیوسته با کولوبومای بزرگ مشیمیه دیده میشود.
حدت بینایی از عدم درک نور تا طبیعی متغیر است. در کلوبومای محدود به عنبیه، بینایی اغلب حفظ میشود. درگیری لکه زرد یا عصب بینایی معمولاً منجر به حدت بینایی ضعیف میشود.
علت اصلی کولوبوما، بسته نشدن شکاف جنینی است.
شکاف جنینی (شکاف جام بینایی) در هفته چهارم جنینی تشکیل شده و در هفته پنجم تکمیل میشود. بسته شدن آن از هفته ششم شروع شده و در هفته هفتم به پایان میرسد. اگر این فرآیند بسته شدن به هر دلیلی مختل شود، کولوبوما ایجاد میشود. نقش ویتامین A نیز در این زمینه مطرح شده است.
چندین ژن در ایجاد کلوبوما نقش دارند.
| ژن | بیماری/فنوتیپ مرتبط |
|---|---|
| PAX2 | سندرم کلیه-کولوبوما 5) |
| CHD7 | سندرم CHARGE |
| FZD5 | OC علامتدار + میکروقرنیه6) |
| TENM3 | MCOPS15 (میکروکورنئا + تاخیر رشدی) 8) |
| FAT1 | کولوبوما + نفروپاتی9) |
| YAP1 | مرتبط با کولوبوما |
| ABCB6 | مرتبط با کولوبوما |
| SALL2 | مرتبط با کولوبوما |
کولوبوما ممکن است با سندرمهای سیستمیک زیر همراه باشد.
این یک سندرم ناهنجاریهای متعدد است که در اثر جهش در ژن CHD7 ایجاد میشود. نام آن از حروف اول کولوبوما (C)، بیماری قلبی (H)، آترزی choanal (A)، تأخیر رشد و تکامل (R)، هیپوپلازی تناسلی (G) و ناهنجاری گوش (E) گرفته شده است و بر اساس ترکیبی از این یافتهها تشخیص داده میشود.
آزمایشهای جامع ژنتیکی مانند توالییابی کامل اگزوم (WES) انجام میشود، اما میزان تشخیص کمتر از ۳۰٪ باقی میماند6).
کولوبوما بسته به محل آن نیاز به افتراق از بیماریهای زیر دارد.
| محل | تشخیصهای افتراقی اصلی |
|---|---|
| پلک | سندرم بند آمنیوتیک، تروما |
| عنبیه | آنیریدیا، جداشدگی تروماتیک عنبیه |
| عصب بینایی | سندرم گل صبحگاهی، هیپوپلازی عصب بینایی |
هیچ درمان قطعی برای کولوبوم وجود ندارد و درمان عمدتاً بر اساس درمان علامتی و مدیریت عوارض متناسب با ناحیه درگیر است.
Castilla-Martinez و همکاران (2024) در یک مورد کولوبومای عنبیه، عدسی و زونولهای Zinn همراه با آب مروارید، از جراحی آب مروارید با لیزر فمتوثانیه (FLACS) همراه با مردمکسازی و کارگذاری حلقه کششی کپسول (CTR) استفاده کردند. دید پس از عمل به logMAR 0.2 بهبود یافت4).
در کلوبوم عصب بینایی، به دلیل ناهنجاری در صفحه کریبریفورم، شریان و ورید مرکزی شبکیه در پشت دیسک بینایی منشعب میشوند و عروق شبکیه از چندین نقطه در حاشیه دیسک بینایی منشأ میگیرند. در زیر دیسک بینایی، اغلب آتروفی کوریورتینال ناشی از بسته نشدن شکاف جنینی دیده میشود.
برای جداشدگی شبکیه از نوع سوراخدار، ویترکتومی انجام میشود. تکنیکهای جراحی مانند بازگرداندن شبکیه با استفاده از چسب فیبرین 7) و فتوکواگولاسیون داخل چشمی همراه با تامپوناد گازی 3) گزارش شدهاند. در موارد جداشدگی سروزی شبکیه، ممکن است بهبود خودبهخودی رخ دهد و برنامه درمانی به صورت فردی تعیین میشود.
کاپ بینایی در هفته چهارم جنینی از اکتودرم عصبی تشکیل میشود. در سمت شکمی کاپ بینایی، شکاف جنینی (شکاف کاپ بینایی) ایجاد میشود که شریان زجاجیه از آن عبور میکند. این شکاف در هفته پنجم کامل میشود و بسته شدن آن از هفته ششم آغاز میگردد. بسته شدن از نزدیک ناحیه استوا شروع شده و به سمت جلو (سمت عنبیه) و عقب (سمت عصب بینایی) پیش میرود و در هفته هفتم کامل میشود.
فرآیند بسته شدن شامل تبدیل اپیتلیال به مزانشیمی (EMT) است. سلولهای اپیتلیال عصبی شبکیه در لبه شکاف جنینی، غشای پایه را تجزیه کرده و با کسب ویژگیهای مزانشیمی، با یکدیگر همجوشی میکنند. اختلال در این فرآیند منجر به بروز کلوبوما میشود.
ژن FZD5 گیرنده مسیر سیگنالدهی Wnt را کد میکند. جهشهای کاهشدهنده عملکرد FZD5 باعث ناهنجاری در سیگنال Wnt شده و منجر به بسته نشدن شکاف جنینی و میکروکورنئا میشود6).
سلولهای تاج عصبی (NCC) نیز در ایجاد کلوبوما نقش دارند. NCCها به بافت مزانشیمی اطراف جام بینایی تمایز مییابند و نقش مهمی در فرآیند بسته شدن شکاف جنینی ایفا میکنند2). اختلال در مهاجرت NCC منجر به ناهنجاریهای رشدی عنبیه و مشیمیه میشود.
Cortes-Gonzalez و همکاران (2024) گزارش کردند که جهش missense هموزیگوت در FZD5 (p.M160V) باعث کولوبومای علامتدار چشم و میکروقرنیه میشود6). این جهش الگوی توارث مغلوب را نشان میدهد و تحلیل عملکردی تأیید کرد که فعالسازی وابسته به لیگاند مسیر سیگنالدهی Wnt مختل میشود. میزان تشخیص ژنتیکی کولوبوما کمتر از 30% است و شناسایی ژنهای جدید عامل بیماری میتواند به بهبود تشخیص کمک کند.
ژو و همکاران (2022) گزارش کردند که جهشهای هتروزیگوت مرکب در ژن TENM3 باعث MCOPS15 (میکروقرنیه، کولوبومای عنبیه و مشیمیه، تأخیر رشد عمومی) میشود8). TENM3 یک پروتئین تراغشایی را کد میکند که در چسبندگی سلولی و رشد عصبی نقش دارد.
Esmaeilzadeh و همکاران (2022) گزارش کردند که جهش در ژن FAT1 در یک خانواده ایرانی مبتلا به کولوبومای عنبیه و نفروپاتی شناسایی شده است9). FAT1 عضوی از ابرخانواده کادهرینها است که در قطبیت سلولی و مورفوژنز بافت نقش دارد.
هو و همکاران (2024) گزارش کردند که جهش تغییر چارچوب c.76delG در ژن PAX2 در یک خانواده مبتلا به گلومرولواسکلروز سگمنتال کانونی (FSGS) شناسایی شده است5). این یافته نشان میدهد که طیف فنوتیپی سندرم کولوبومای کلیوی گستردهتر از آنچه قبلاً تصور میشد است.
Jain و همکاران (2024) یک مورد از جداشدگی شبکیه مرتبط با کولوبوما را گزارش کردند که با استفاده از چسب فیبرین و عمل جااندازی شبکیه درمان شد7). این روش شامل اعمال چسب فیبرین در اطراف پارگی شبکیه در لبه کولوبوما برای تقویت چسبندگی است و در نهایت دید نهایی به 20/50 بهبود یافت.
Ratra و همکاران (2023) موردی از کولوبومای غیرمعمول کوروئید همراه با فیستول اسکلرال پس از تروما را با موفقیت با ویترکتومی + فوتوکواگولاسیون داخل چشمی + تامپوناد گازی درمان کردند3).
Scemla و همکاران (2021) موردی از یک مرد 19 ساله با کلوبوم کوروئید را گزارش کردند که در محل آن فیلتراسیون از طریق صلبیه رخ داده و منجر به افت فشار چشم (4 میلیمتر جیوه) شده بود1). میکروسکوپ اولتراسوند بیومیکروسکوپی نقص صلبیه را تأیید کرد. پس از 6 هفته بهبود خودبهخودی رخ داد و فشار چشم 11 میلیمتر جیوه و حدت بینایی 1.0 حفظ شد.