سندرم آیکاردی (Aicardi syndrome) یک بیماری نادر مادرزادی است که اولین بار در سال ۱۹۶۷ توسط متخصص مغز و اعصاب فرانسوی ژان آیکاردی توصیف شد. وراثت آن وابسته به X غالب فرض میشود و تقریباً همه بیماران دختر هستند. در پسران به دلیل کشنده بودن حالت هموزیگوت، تنها موارد نادر با کاریوتایپ XXY (سندرم کلاینفلتر) گزارش شده است.
میزان بروز حدود ۱ در ۱۱۰,۰۰۰ تولد تخمین زده میشود و تعداد مبتلایان در سراسر جهان حدود ۴۰۰۰ نفر است1). همه موارد جهش de novo هستند و هیچ مورد انتقال از والد به فرزند گزارش نشده است؛ خطر عود در خواهر و برادر کمتر از ۱٪ است1).
سهگانه کلاسیک شامل موارد زیر است1):
اسپاسم نوزادی (infantile spasms): معمولاً در حدود ۳ تا ۴ ماهگی شروع میشود.
حفرههای کوروئید-شبکیه (chorioretinal lacunae): ضایعات دایرهای دوطرفه در فوندوس چشم. یافتهای اختصاصی برای این بیماری.
عدم تشکیل جسم پینهای (agenesis of the corpus callosum): فقدان جزئی یا کامل.
پیشآگهی ضعیف است. میانگین سن بقا ۱۸ سال است و احتمال بقا تا ۲۷ سال ۰.۶۲٪ گزارش شده است1).
Qآیا سندرم آیکاردی در پسران نیز رخ میدهد؟
A
این بیماری تقریباً منحصراً در دختران رخ میدهد. تصور میشود که وراثت وابسته به X غالب باشد و در پسران به دلیل هموزیگوت بودن کشنده است. با این حال، چند مورد در پسران با کاریوتایپ XXY (سندرم کلاینفلتر) در جهان گزارش شده است.
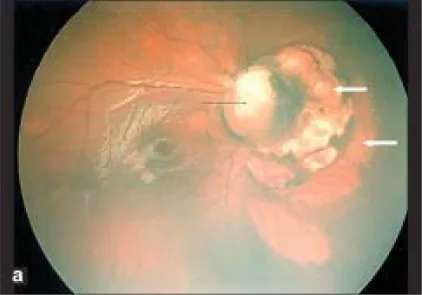
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
عکس Retcam از چشم راست، کولوبومای دیسک بینایی (فلش سیاه) و نواحی گنبدی شکل رنگ پریده با مرزهای تیز در سمت بینی دیسک بینایی را نشان میدهد که نشاندهنده حفرههای کوروئید-شبکیه است (فلشهای سفید).
اولین علامت این بیماری معمولاً اسپاسم نوزادی (infantile spasms) در حدود ۳-۴ ماهگی است. صرع اغلب به مقاوم به درمان تبدیل میشود و انواع مختلف تشنج را شامل میشود.
تشنج: با اسپاسم نوزادی شروع میشود و به مقاوم به درمان پیشرفت میکند. در یک مورد، تشنج عمومی در ۴ ماهگی ۳-۴ بار در روز (هر بار ۲۰-۲۵ دقیقه) رخ داد1). در مورد دیگر، تشنج مکرر پلک زدن از ۱ ماهگی مشاهده شد2).
تأخیر رشد روانی-حرکتی: همراه با ناتوانی ذهنی شدید، اغلب حرکت مستقل و کسب زبان دشوار است.
اختلال عملکرد دستگاه گوارش: علائم گوارشی مانند یبوست در بیش از ۹۰٪ موارد وجود دارد1).
اختلال بینایی: کاهش بینایی ناشی از ضایعات فوندوس، عدم تشکیل جسم پینهای و ناهنجاریهای قشر مغز.
در میان یافتههای چشمی این بیماری، لاکونای کوریورتینال (choroidal-retinal lacuna) یک یافته پاتوگنومونیک (pathognomonic) برای این بیماری محسوب میشود.
لاکونای کوریورتینال
توزیع: دوطرفه. در اطراف دیسک بینایی و قطب خلفی متراکم است، اما به نواحی محیطی نیز گسترش مییابد.
ظاهر: ضایعات گرد تا بیضی شکل به رنگ زرد-سفید تا صورتی. شیوع ۷۰ تا ۹۰٪1).
بافتشناسی: فقدان اپیتلیوم رنگدانهدار شبکیه (RPE) که از لایه کوروئید تا لایه صلبیه برهنه امتداد دارد2).
سیر: ممکن است اندازه و تعداد ضایعات پس از جراحی به مرور زمان افزایش یابد2).
سایر یافتههای چشمی
کولوبوم عصب بینایی: در حدود ۴۴٪ موارد همراه است.
میکروفتالمی: در حدود ۲۰٪ موارد مشاهده میشود.
شبکیه بدون عروق محیطی: ممکن است یک ناحیه بدون عروق ۳۶۰ درجه تشکیل شود2).
جداشدگی شبکیه کششی (TRD): ممکن است همراه با بافت ساقهای عروقی (stalk tissue) رخ دهد2).
در گزارشهای موردی، نمونهای با خونریزی پیششبکیهای و ناحیه بدون عروق محیطی ۳۶۰ درجه در چشم راست، و بافت ساقهای (ساقه فیبروواسکولار) و جداشدگی شبکیه کششی در چشم چپ ثبت شده است2). همچنین میتواند باعث اختلال بینایی قشری (cortical visual impairment; CVI) شود3).
آژنزی جسم پینهای: فقدان کامل یا جزئی جسم پینهای در تمام موارد وجود دارد1). نازک شدن جسم پینهای (dysgenesis) نیز به عنوان یک نوع تغییر گزارش شده است2).
ناهنجاری قشر مغز: پلیمیکروژیری (polymicrogyria، نوع 2 طبقهبندی بارکوویچ)، ندولهای ماده خاکستری اطراف بطنی، و کیستهای چندحفرهای در MRI تأیید میشوند1).
یافتههای EEG: الگوی مشخصی از ریتم پلیمورفیک با دامنه بالا همراه با تخلیههای چندکانونی اسپایک و موج را نشان میدهد1).
ناهنجاریهای اسکلتی: در 40 تا 60٪ موارد، جوش خوردن مهرههای سینهای (T9-T10) و مهره پروانهای (T8) مشاهده میشود1).
Qآیا لکونهای کوریورتینال در طول زمان تغییر میکنند؟
A
ممکن است پیشرفت کنند. گزارش شده است که پس از مداخله جراحی چشم، لکونها جدیداً قابل مشاهده میشوند یا اندازه و تعداد آنها در طول زمان افزایش مییابد2). پیگیری منظم با معاینات فوندوسکوپی مهم است.
سندرم ایکاردی احتمالاً به صورت وابسته به X غالب به ارث میرسد، اما ژن عامل تاکنون شناسایی نشده است1). همه موارد جهشهای de novo هستند و اساساً موارد خانوادگی دیده نمیشود. خطر عود برای خواهر و برادر کمتر از 1٪ است و توصیه میشود پس از مشاوره ژنتیک، برنامهریزی برای فرزند بعدی بررسی شود1).
در گزارشهای موردی اخیر، جهش در ژن TREX1 (c.292_293insA، p.(Cys99Metfs)) در یک مورد شناسایی شده است2). TREX1 روی کروموزوم 3 قرار دارد که تا حدی با فرضیه وابسته به X در تضاد است، بنابراین مکان ژن عامل همچنان مورد بحث است.
الگوی وراثت: وابسته به X غالب (فرضی). مردان هموزیگوت در دوره جنینی کشنده هستند.
ماهیت جهش: همه جهشهای de novo. وراثتی (انتقال از والد به فرزند) وجود ندارد.
خطر عود: خطر عود برای خواهر و برادر کمتر از 1٪ است1).
همکاری چندتخصصی: تشخیص نیازمند همکاری متخصصان مغز و اعصاب، چشم، ارتوپد و ژنتیک است 1).
Qآیا آزمایش ژنتیکی میتواند تشخیص قطعی دهد؟
A
در حال حاضر، هیچ ژن عامل مشخصی برای تشخیص قطعی شناسایی نشده است، بنابراین تشخیص تنها با آزمایش ژنتیکی امکانپذیر نیست 1). تشخیص عمدتاً بالینی و بر اساس ترکیبی از علائم بالینی، یافتههای فوندوس و تصویربرداری است. پیشرفت در آنالیز ژنومی جامع، امید به شناسایی ژن عامل در آینده را افزایش میدهد.
پس از ویترکتومی، بهبود رشد طول محوری چشم تأیید شده است؛ به طوری که طول محوری چشم چپ از 17.45 میلیمتر در یک ماهگی به 24.41 میلیمتر در 26 ماهگی افزایش یافته است 2). همچنین مواردی گزارش شده است که پس از جراحی، لاکونای کوریورتینال جدید قابل مشاهده شده و به تشخیص قطعی کمک کرده است 2).
Qآیا درمان جراحی چشمی امکانپذیر است؟
A
بله، امکانپذیر است. فتوکوآگولاسیون لیزری برای شبکیه بدون عروق محیطی و ویترکتومی 23G برای جداشدگی کششی شبکیه ممکن است مؤثر باشند 2). گزارشهایی از تسریع رشد کره چشم پس از جراحی وجود دارد. با این حال، تعداد موارد کم است و نیاز به مدیریت دقیق در مراکز تخصصی دارد.
جهش de novo روی کروموزوم X به عنوان علت این بیماری فرض میشود. الگوی غیرفعالسازی کروموزوم X (لیونیزاسیون) باعث تنوع فنوتیپی در همان جهش میشود. در مردان، به دلیل هموزیگوت بودن، در دوره جنینی کشنده است و تنها مردان با کاریوتایپ XXY میتوانند زنده بمانند.
اخیراً، جهش ژن TREX1 (c.292_293insA, p.(Cys99Metfs)) در یک بیمار تشخیص داده شده با این بیماری یافت شده است 2). از آنجایی که TREX1 روی کروموزوم 3 قرار دارد، ممکن است با فرضیه وابسته به X تناقض داشته باشد و احتمال وجود ژنهای عامل غیر وابسته به X را مطرح کند.
از نظر بافتشناسی، لاکونای کوریورتینال با فقدان اپیتلیوم رنگدانهدار شبکیه (RPE) از لایه مویرگهای کوروئید تا صلبیه برهنه مشخص میشود 2). در ناحیه缺损، اپیتلیوم رنگدانهدار شبکیه و لایه مویرگهای کوروئید وجود ندارند و دیسپلازی شبکیه عصبی تمایزنیافته دیده میشود.
باقیماندگان عروق جنینی پایدار (persistent fetal vasculature) در تشکیل ساقه فیبروواسکولار (stalk tissue) و شبکیه بدون عروق محیطی نقش دارند 2). این عروق غیرطبیعی باعث جداشدگی کششی شبکیه میشوند.
اختلال بینایی قشری (CVI) به عنوان کاهش عملکرد بینایی اکتسابی ناشی از ناهنجاریهای ساختاری مغز مانند پلیمیکروژیری، هتروتوپی ماده خاکستری و آژنزی جسم پینهای ایجاد میشود 3).
ناهنجاریهای مغزی (پلیمیکروژیری، آژنزی جسم پینهای) بر اساس اختلال مهاجرت سلولهای عصبی در دوره جنینی است. مکانیسم مولکولی زمینهساز این اختلال مهاجرت هنوز ناشناخته است و با شناسایی ژن عامل، تحقیقات آینده مورد انتظار است.
7. تحقیقات جدید و چشمانداز آینده (گزارشهای مرحله تحقیقاتی)
ژن عامل هنوز ناشناخته است، اما تشخیص جهش در ژن TREX1 2) سرنخ جدیدی است که نشان میدهد جهشهای غیر وابسته به X ممکن است درگیر باشند. با گسترش آنالیز ژنومی جامع (WES و WGS)، انتظار میرود شناسایی ژن عامل تسریع شود. تأیید فرضیه وابسته به X و غیر وابسته به X یک چالش اصلی در آینده است.
گزارشهایی از انواع جهشیافته که در آنها فقدان کامل جسم پینهای وجود ندارد و فقط نازکشدگی (dysgenesis) دیده میشود، در حال افزایش است 2). حتی در مواردی که سهگانه کامل وجود ندارد، با معاینه دقیق فوندوس و استفاده از معیارهای تشخیصی گسترده، ممکن است دقت تشخیص بهبود یابد.
Kang و همکاران (2022) گزارش کردند که در یک مورد ویتئورتینوپاتی دوطرفه در سندرم آیکاردی، لیزر فوتوکوآگولاسیون و ویترکتومی 23G انجام دادند که به حفظ رشد کره چشم و محافظت از عملکرد بینایی کمک کرد 2). همچنین ثبت شد که پس از جراحی، لاکونای کوریورتینال جدید قابل مشاهده شد، که نشان میدهد جراحی چشم میتواند به تشخیص قطعی نیز کمک کند.
آگاهی در حال افزایش است که غربالگری زودهنگام فوندوس و مداخله سریع برای محافظت از عملکرد شبکیه مهم است 2). لیزر به مناطق بدون عروق محیطی ممکن است از پیشرفت رتینوپاتی پرولیفراتیو جلوگیری کند و جمعآوری موارد بیشتر در آینده مطلوب است.
تحقیقات در مورد استفاده از کانابیدیول (CBD) و رژیم کتوژنیک برای کنترل صرع در حال پیشرفت است 1). انتظار میرود گزینههای درمانی جدیدی برای صرع مقاوم به درمان ایجاد شود.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.