Das Aicardi-Syndrom ist eine angeborene seltene Erkrankung, die erstmals 1967 von dem französischen Neurologen Jean Aicardi beschrieben wurde. Es wird als X-chromosomal-dominant vererbt und betrifft fast ausschließlich Mädchen. Bei Jungen ist es im hemizygoten Zustand letal, es gibt nur wenige Berichte bei Jungen mit XXY-Karyotyp (Klinefelter-Syndrom).
Die Inzidenz wird auf etwa 1:110.000 Geburten geschätzt, weltweit sind etwa 4.000 Menschen betroffen 1). Alle Fälle sind De-novo-Mutationen, es gibt keine Übertragung von Eltern auf Kinder, und das Wiederholungsrisiko für Geschwister liegt unter 1 % 1).
Die klassische Trias umfasst die folgenden drei 1):
Blitz-Nick-Salaam-Krämpfe (Infantile Spasmen): Beginn im Alter von etwa 3–4 Monaten.
Chorioretinale Lakunen (chorioretinal lacunae) : bilaterale runde Fundusläsionen. Für diese Erkrankung spezifischer Befund.
Agenesie des Corpus callosum (agenesis of the corpus callosum) : teilweises oder vollständiges Fehlen.
Die Prognose ist schlecht. Das durchschnittliche Überlebensalter beträgt 18 Jahre, und die Wahrscheinlichkeit, bis zum Alter von 27 Jahren zu überleben, wird mit 0,62 % angegeben 1).
QTritt das Aicardi-Syndrom auch bei Jungen auf?
A
Diese Erkrankung tritt fast ausschließlich bei Mädchen auf. Es wird ein X-chromosomal dominanter Erbgang vermutet, da sie bei hemizygoten Jungen letal wäre. Allerdings gibt es weltweit einige wenige Berichte über Jungen mit XXY-Karyotyp (Klinefelter-Syndrom).
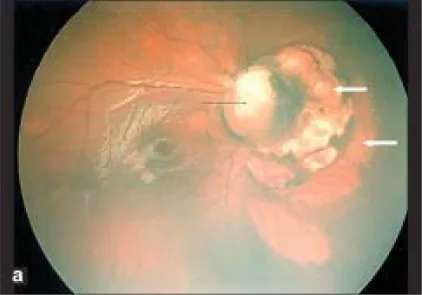
Parag K Shah; V Narendran; N Kalpana. Aicardi syndrome: The importance of an ophthalmologist in its diagnosis. Indian J Ophthalmol. 2009 May-Jun; 57(3):234-236 Figure 1. PMCID: PMC2683450. License: CC BY.
Retcam-Foto des rechten Auges zeigt ein Optikusdiskuskolobom (schwarzer Pfeil) und kuppelförmige blasse Areale mit scharfen Grenzen nasal des Optikusdiskus, die auf chorioretinale Lakunen hindeuten (weiße Pfeile).
Das Erstsymptom dieser Erkrankung sind in der Regel BNS-Krämpfe (Blitz-Nick-Salaam-Krämpfe), die im Alter von etwa 3-4 Monaten auftreten. Die Epilepsie entwickelt sich häufig zu einer medikamentenresistenten Form und geht mit verschiedenen Anfallstypen einher.
Epileptische Anfälle : beginnen als BNS-Krämpfe und entwickeln sich zu medikamentenresistenter Epilepsie. In einem Fall traten im Alter von 4 Monaten generalisierte Krämpfe 3-4 Mal täglich (jeweils 20-25 Minuten anhaltend) auf 1). In einem anderen Fall wurden ab dem 1. Lebensmonat häufige Blinzelanfälle beobachtet 2).
Psychomotorische Entwicklungsverzögerung : mit schwerer geistiger Behinderung, oft mit Schwierigkeiten bei unabhängiger Fortbewegung und Spracherwerb.
Gastrointestinale Funktionsstörung : Verdauungssymptome wie Verstopfung treten bei über 90 % der Fälle auf 1).
Sehbehinderung : verursacht durch Fundusläsionen, Corpus-callosum-Agenesie und kortikale Fehlbildungen.
Unter den ophthalmologischen Befunden dieser Erkrankung gelten Aderhaut-Netzhaut-Lakunen als pathognomonisch.
Aderhaut-Netzhaut-Lakunen
Verteilung : beidseitig. Dicht um die Papille und am hinteren Pol, aber auch in der Peripherie.
Aussehen : runde bis ovale, gelb-weiße bis rosafarbene Läsionen. Prävalenz 70–90 %1).
Histologie : Defekt des retinalen Pigmentepithels (RPE), der von der Aderhautschicht bis zur freiliegenden Sklera reicht2).
Verlauf : Größe und Anzahl können nach einer Operation im Laufe der Zeit zunehmen2).
Andere Augenveränderungen
Optikuskolobom : tritt bei etwa 44 % der Fälle auf.
Mikrophthalmus : wird bei etwa 20 % beobachtet.
Periphere avaskuläre Netzhaut : Es kann sich eine 360-Grad-avaskuläre Zone bilden2).
Traktionsamotio retinae (TRD) : kann in Verbindung mit Stielgewebe (stalk tissue) auftreten2).
In Fallberichten wurde ein Beispiel mit einer präretinalen Blutung und einer 360-Grad-peripheren avaskulären Zone im rechten Auge sowie Stielgewebe (fibrovaskulärer Stiel) und Traktionsamotio im linken Auge dokumentiert2). Es kann auch eine Ursache für kortikale Sehbeeinträchtigung (CVI) sein3).
Balkenmangel : partielle oder vollständige Agenseie des Corpus callosum ist in allen Fällen vorhanden1). Eine Dysgenese (Verdünnung) des Corpus callosum wurde ebenfalls als Variante berichtet2).
Großhirnrindenfehlbildung: Polymikrogyrie (Barkovich-Klassifikation Typ 2), periventrikuläre graue Substanz-Knoten und multilokuläre Zysten werden im MRT nachgewiesen1).
EEG-Befund: Zeigt ein charakteristisches Muster mit hochamplitudigen polymorphen Rhythmen und multifokalen Spike-Wave-Entladungen1).
Skelettanomalien: Bei 40–60 % finden sich Fusionen der Brustwirbel (T9–T10) oder Schmetterlingswirbel (T8)1).
QVerändern sich die Aderhaut-Netzhaut-Lakunen im Laufe der Zeit?
A
Sie können fortschreiten. Es wurde berichtet, dass nach ophthalmologischen chirurgischen Eingriffen neue Lakunen sichtbar werden oder dass Größe und Anzahl im Laufe der Zeit zunehmen2). Eine regelmäßige Nachsorge mittels Funduskopie ist wichtig.
Das Aicardi-Syndrom wird als X-chromosomal-dominant vererbt angenommen, aber das verantwortliche Gen wurde bisher nicht identifiziert1). Alle Fälle sind De-novo-Mutationen, eine familiäre Häufung wird grundsätzlich nicht beobachtet. Das Wiederholungsrisiko für Geschwister liegt unter 1 %, und es wird empfohlen, nach genetischer Beratung die Planung eines weiteren Kindes zu erwägen1).
In einem aktuellen Fallbericht wurde bei einem Patienten eine TREX1-Genmutation (c.292_293insA, p.(Cys99Metfs)) nachgewiesen2). TREX1 liegt auf Chromosom 3, was der X-chromosomalen Hypothese teilweise widerspricht, und die Lokalisierung des verantwortlichen Gens wird weiterhin diskutiert.
Erbgang: X-chromosomal-dominant (angenommen). Hemizygote Männer sind letal in utero.
Art der Mutation: Alle De-novo-Mutationen. Es gibt keine Vererbung (von Eltern auf Kinder).
Die Diagnose des Aicardi-Syndroms basiert hauptsächlich auf der klinischen Diagnose, da das verantwortliche Gen nicht identifiziert ist. Die Bestätigung der folgenden klassischen Trias steht im Mittelpunkt der Diagnose1).
Infantile Spasmen (West-Syndrom)
Chorioretinale Lakunen (chorioretinal lacunae)
Agenesie des Corpus callosum (agenesis of the corpus callosum)
Auch wenn nur zwei der drei Merkmale erfüllt sind, kann die Diagnose nach den 1999 festgelegten erweiterten Diagnosekriterien gestellt werden.
Die Zusammensetzung der erweiterten Diagnosekriterien ist unten dargestellt.
Klassifikation
Hauptpunkte
Hauptmerkmale
Optikuskolobom, kortikale Fehlbildung, Heterotopie der grauen Substanz, intrakranielle Zyste, Plexus-choroideus-Papillom
MRT (Kopf) : Bestätigung der Corpus-callosum-Agenesie. Beurteilung von Polymikrogyrie, Heterotopie der grauen Substanz, Erweiterung der Seitenventrikel, bilaterale thalamische Streifenzysten, Hippocampushypoplasie usw.1)2)
EEG (Elektroenzephalogramm) : Bestätigung eines hochvoltmorphischen Rhythmus und eines Musters multifokaler Spike-Wave-Entladungen.1)
Fundusuntersuchung: Bestätigung des Vorhandenseins von chorioretinalen Lakunen. Die Fluoreszenzangiographie (FA) ist zur Beurteilung avaskulärer Bereiche nützlich 2).
Röntgen-Thorax (AP): Bestätigung von Skelettanomalien (Fusionswirbel, Schmetterlingswirbel) 1).
Multidisziplinäre Zusammenarbeit: Die Diagnose erfordert die Zusammenarbeit von Neurologie, Augenheilkunde, Orthopädie und Genetik 1).
QKann ein Gentest die Diagnose bestätigen?
A
Derzeit wurde kein ursächliches Gen für eine definitive Diagnose identifiziert, daher kann allein ein Gentest die Diagnose nicht bestätigen 1). Die klinische Diagnose basiert auf der Kombination von klinischen Symptomen, Fundusbefunden und Bildgebungsbefunden. Fortschritte in der umfassenden Genomanalyse lassen hoffen, dass das ursächliche Gen in Zukunft identifiziert wird.
Es gibt keine kurative Behandlung. Die Behandlungsziele basieren auf drei Säulen: Anfallskontrolle, Behandlung ophthalmologischer Komplikationen und entwicklungsfördernde Rehabilitation.
Epilepsiemanagement
Mittel der ersten Wahl: Phenytoin, Levetiracetam, Clobazam usw. 1).
Therapierefraktäre Fälle: Cannabidiol (CBD), ketogene Diät, Corpus-callosum-Durchtrennung, Vagusnervstimulation können versucht werden 1).
Augenärztlicher Eingriff
Laserphotokoagulation: Durchführung an der peripheren avaskulären Netzhaut, um das Fortschreiten der proliferativen Retinopathie zu verhindern 2).
Vitrektomie: 23G-Vitrektomie bei traktiver Netzhautablösung (TRD) 2).
Rehabilitation
Früher Beginn: Physiotherapie, Ergotherapie, Sprachtherapie und Sehtherapie unmittelbar nach der Diagnose beginnen 1).
Low-Vision-Versorgung: Bereitstellung von Sehhilfen und Umgebungsanpassungen bei Sehbehinderungen.
Es werden Beispiele für chirurgische Eingriffe bei ophthalmologischen Komplikationen vorgestellt.
Auge
Befund
Behandlung
Rechtes Auge
Präretinale Blutung, avaskuläre Zone 360°
Laserphotokoagulation
Linkes Auge
Stielgewebe, TRD
23G-Vitrektomie
Nach der Vitrektomie wurde eine Erholung des axialen Längenwachstums bestätigt: Die axiale Länge des linken Auges, die im Alter von 1 Monat 17,45 mm betrug, wuchs bis zum Alter von 26 Monaten auf 24,41 mm an 2). Darüber hinaus wurde über Fälle berichtet, in denen nach der Operation neu choroidale Netzhautlakunen sichtbar wurden, was zur Diagnosesicherung beitrug 2).
QIst eine ophthalmologische Operation möglich?
A
Ja, das ist möglich. Eine Laserphotokoagulation der peripheren avaskulären Netzhaut oder eine 23G-Vitrektomie bei traktiver Netzhautablösung kann in einigen Fällen wirksam sein 2). Es gibt auch Berichte über ein beschleunigtes Augenwachstum nach der Operation. Allerdings ist die Fallzahl gering, und eine sorgfältige Behandlung in einer spezialisierten Einrichtung ist erforderlich.
Eine De-novo-Mutation auf dem X-Chromosom wird als Ursache dieser Krankheit angenommen. Durch das Muster der X-Chromosom-Inaktivierung (Lyonisation) kann dieselbe Mutation zu phänotypischer Vielfalt führen. Bei Männern ist die Mutation im hemizygoten Zustand während der Embryonalphase letal, nur Männer mit einem XXY-Karyotyp können überleben.
In letzter Zeit wurde bei einem Patienten mit dieser Diagnose eine TREX1-Genmutation (c.292_293insA, p.(Cys99Metfs)) nachgewiesen 2). Da TREX1 auf Chromosom 3 liegt, könnte dies der X-chromosomalen Hypothese widersprechen und auf die Möglichkeit nicht X-chromosomaler ursächlicher Gene hinweisen.
Histologisch zeigen chorioretinale Lakunen einen Defekt des retinalen Pigmentepithels (RPE), der von der Choriokapillaris bis zur nackten Sklera reicht 2). In den Defektbereichen fehlen RPE und Choriokapillaris, und es findet sich eine Dysplasie der undifferenzierten neurosensorischen Netzhaut.
Es wird angenommen, dass Reste des persistierenden fetalen Gefäßsystems (persistent fetal vasculature) an der Bildung von fibrovaskulärem Stielgewebe und der peripheren avaskulären Netzhaut beteiligt sind 2). Diese abnormalen Gefäße verursachen eine traktive Netzhautablösung.
Die kortikale Sehbeeinträchtigung (CVI) entsteht als erworbene Verschlechterung der Sehfunktion aufgrund von Hirnstrukturanomalien wie Polymikrogyrie, ektoper grauer Substanz und Balkenmangel 3).
Hirnfehlbildungen (Polymikrogyrie, Balkenmangel) beruhen auf einer Störung der neuronalen Migration während der Embryonalphase. Der molekulare Mechanismus dieser Migrationsstörung ist noch ungeklärt, und zukünftige Forschung wird zusammen mit der Identifizierung des ursächlichen Gens erwartet.
7. Aktuelle Forschung und Zukunftsaussichten (Berichte aus der Forschungsphase)
Das verursachende Gen bleibt unbekannt, aber der Nachweis von TREX1-Genmutationen 2) ist ein neuer Hinweis, der auf eine mögliche Beteiligung nicht-X-chromosomaler Mutationen hindeutet. Die Verbreitung umfassender Genomanalysen (WES/WGS) wird voraussichtlich die Identifizierung des verursachenden Gens beschleunigen. Die Überprüfung der X-chromosomalen und nicht-X-chromosomalen Hypothesen ist eine zukünftige Hauptaufgabe.
Erkennung von Varianten (atypischen Formen) des Aicardi-Syndroms
Es häufen sich Berichte über Varianten, die keine vollständige Balkenagenesie, sondern nur eine Verdünnung (Dysgenesie) des Corpus callosum zeigen 2). Auch bei Fällen, die nicht alle drei Kriterien erfüllen, könnte eine detaillierte Fundusuntersuchung und die Anwendung erweiterter Diagnosekriterien die Diagnosegenauigkeit verbessern.
Kang et al. (2022) berichteten über die Durchführung einer Laserphotokoagulation und 23G-Vitrektomie bei einem Fall von bilateraler Vitreoretinopathie beim Aicardi-Syndrom, was zur Aufrechterhaltung des Augenwachstums und zum Schutz der Sehfunktion beitrug 2). Sie dokumentierten auch, dass nach der Operation neu chorioretinale Lakunen sichtbar wurden, was zeigt, dass eine Augenoperation auch zur Diagnosesicherung beitragen kann.
Die Bedeutung eines frühen Fundus-Screenings und eines schnellen Eingreifens zum Schutz der Netzhautfunktion wird zunehmend anerkannt 2). Laser auf periphere avaskuläre Bereiche könnte das Fortschreiten der proliferativen Retinopathie verhindern, und eine Sammlung von Fällen ist für die Zukunft wünschenswert.
Die Forschung zur Anwendung von Cannabidiol (CBD) und ketogener Diättherapie zur Epilepsiekontrolle schreitet voran 1). Die Etablierung neuer Behandlungsoptionen für therapierefraktäre Epilepsie wird erwartet.
Jakhar S, Yadav D, Bhalla K, Jindal K, Acharya R. Aicardi syndrome: Clinical spectrum of a rare disorder. J Family Med Prim Care. 2025;14:1145-6. doi:10.4103/jfmpc.jfmpc_1065_24. PMID:40256067; PMCID:PMC12007781.
Kang EYC, Chong YJ, Lien R, Wu WC. A rare case of bilateral vitreoretinopathy of Aicardi syndrome. Am J Ophthalmol Case Rep. 2022;26:101467. doi:10.1016/j.ajoc.2022.101467. PMID:35345580; PMCID:PMC8956863.
Melinda Y. Chang, Mark S. Borchert. Advances in the evaluation and management of cortical/cerebral visual impairment in children. Survey of Ophthalmology. 2020;65(6):708-724. doi:10.1016/j.survophthal.2020.03.001.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.