RRMS
Schubförmig-remittierende MS (RRMS) : der häufigste Subtyp. Schübe dauern länger als 24 Stunden, mit vollständiger oder teilweiser Remission zwischen den Schüben.

Multiple Sklerose (MS) ist eine entzündliche demyelinisierende Erkrankung der weißen Substanz des zentralen Nervensystems (ZNS), die zu vielfältigen neurologischen Symptomen mit Schüben und Remissionen führt. Charakteristisch sind vernarbte sklerotische Läsionen durch Gliose, wobei in der Regel nur das ZNS betroffen ist und das periphere Nervensystem ausgespart bleibt.
Die geschätzte Prävalenz in den USA beträgt 1–1,5 pro 1.000 Personen1). Weltweit sind 2,1 Millionen Menschen betroffen, mit einer Häufung in den hohen Breitengraden der Nord- und Südhalbkugel. Das Durchschnittsalter bei Erkrankungsbeginn liegt zwischen 15 und 45 Jahren, das Durchschnittsalter bei Diagnose bei 30 Jahren. Die bevorzugte Altersgruppe ist 15–50 Jahre, Frauen sind häufiger betroffen (Gipfel Ende 20), das Geschlechterverhältnis beträgt 1:2,9.
Die MS hat vier Hauptsubtypen. RRMS (schubförmig-remittierend) beginnt meist im Alter von 25–29 Jahren, SPMS tritt häufiger im Alter von 40–49 Jahren auf1).
RRMS
Schubförmig-remittierende MS (RRMS) : der häufigste Subtyp. Schübe dauern länger als 24 Stunden, mit vollständiger oder teilweiser Remission zwischen den Schüben.
SPMS
Sekundär progrediente MS (SPMS) : Übergang von RRMS. Auch in Remissionsphasen kommt es zu einer fortschreitenden Behinderungszunahme.
PPMS
Primär progrediente MS (PPMS) : von Beginn an fortschreitende Behinderungszunahme ohne Schübe, langsame Progression.
CIS
Klinisch isoliertes Syndrom (CIS) : erste klinische Episode, die zu MS werden kann. Ermöglicht frühen Therapiebeginn.
Die MS wird in vier Subtypen eingeteilt: RRMS (schubförmig-remittierend), SPMS (sekundär progredient), PPMS (primär progredient) und CIS (klinisch isoliertes Syndrom). Am häufigsten ist RRMS mit Schüben und Remissionen. SPMS entwickelt sich aus RRMS, während PPMS von Anfang an progredient ist.
Bei 75% der Patienten ist das erste Symptom eine einzelne Beschwerde, 45% haben motorische/sensorische Symptome und 20% visuelle Symptome.
Augensymptome
Allgemeine neurologische Symptome
Die Exazerbation beginnt akut bis subakut und dauert einige Tage bis mehrere Monate an. In 85 % der Fälle kommt es zu einer Besserung oder zum Verschwinden der Symptome, aber in 10–15 % bleiben Residuen bestehen.
Sie manifestiert sich häufig als einseitige schmerzhafte Sehverschlechterung. Orbitalschmerz tritt bei 92 % auf und wird durch Augenbewegungen verstärkt. Außerdem wird das Uhthoff-Phänomen beobachtet, bei dem ein Anstieg der Körpertemperatur (Baden, Bewegung) die Symptome vorübergehend verschlechtert.
Die genaue Ursache der MS ist unbekannt, aber es wird angenommen, dass ein autoimmuner Mechanismus an der Entstehung beteiligt ist.
Genetische Faktoren sind beteiligt, aber die Konkordanzrate bei eineiigen Zwillingen beträgt nur 25–30 %. Obwohl HLA-Polymorphismus und über 100 Risikoloci identifiziert wurden, wird angenommen, dass nicht nur die genetische Veranlagung, sondern auch Umweltfaktoren eine wichtige Rolle bei der Entstehung der Krankheit spielen.
Die McDonald-Kriterien von 2017 (Revision 2024) werden verwendet. Der Nachweis der zeitlichen und räumlichen Dissemination (DIT/DIS) von demyelinisierenden Läsionen des zentralen Nervensystems ist grundlegend. In der Revision von 2024 wurde der Sehnerv als fünfte topografische Region hinzugefügt. In Japan gibt es auch die Diagnosekriterien für Multiple Sklerose des Ministeriums für Gesundheit, Arbeit und Soziales von 2015.
Die fünf topografischen Regionen der räumlichen Dissemination (DIS) sind wie folgt:
Nachweis der zeitlichen Dissemination (DIT): ≥2 Schübe, oder gleichzeitiges Vorhandensein von kontrastmittelaufnehmenden und nicht aufnehmenden Läsionen im MRT, neue T2-Läsionen, oder oligoklonale Banden im Liquor 1).
Für die Diagnose einer PPMS sind neben einer Behinderungsprogression ≥1 Jahr mindestens 2 der folgenden Befunde erforderlich: zerebrale T2-Läsionen, spinale T2-Läsionen (≥2), oder oligoklonale Banden im Liquor 1).
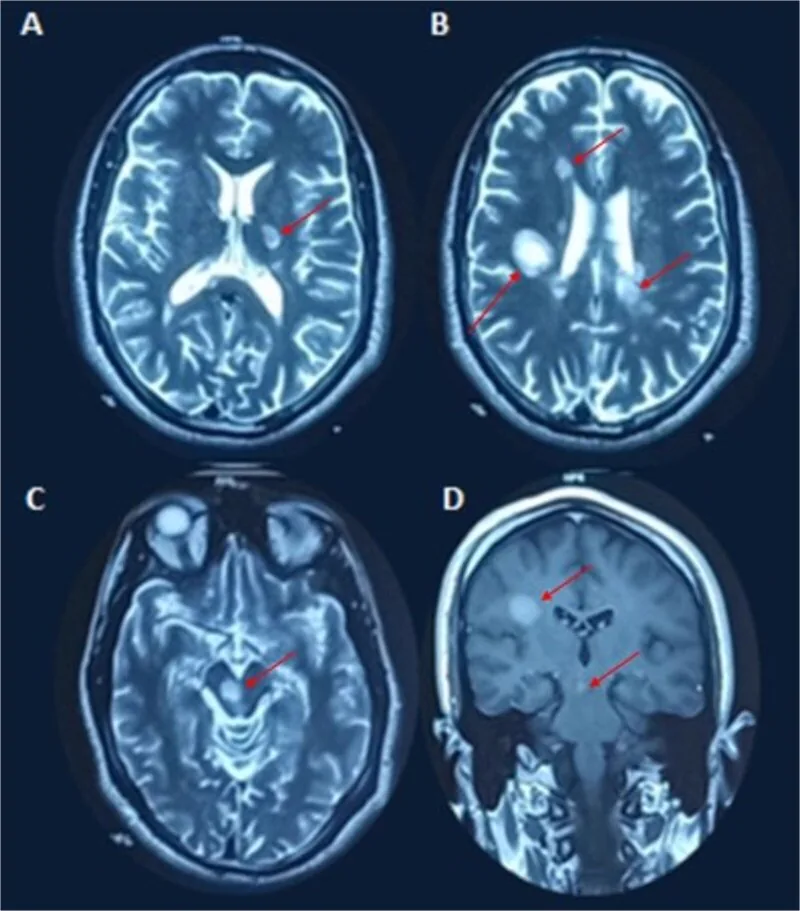
Demyelinisierungsplaques werden als T2-hyperintense Läsionen oder Gadolinium-anreichernde Läsionen nachgewiesen.
Nützlich, wenn das MRT nicht eindeutig ist oder zur Vorhersage des Krankheitsverlaufs 1). Kann frühe, asymptomatische Demyelinisierung vor der MRT-Sichtbarkeit erkennen. Bei 65% zeigen sich Latenzverlängerung und Amplitudenminderung.
Die Abgrenzung zu folgenden Erkrankungen ist wichtig; bei atypischen Fällen werden Zusatzuntersuchungen durchgeführt.
| Erkrankungskategorie | Wichtige Differenzialdiagnosen |
|---|---|
| Demyelinisierende Erkrankungen | NMO (Devic-Krankheit), ADEM, MOGAD |
| Infektiös | Sarkoidose, Tuberkulose, Syphilis, Lyme-Borreliose |
| Autoimmun | SLE, Sjögren-Syndrom, Morbus Behçet |
| Sehnervenerkrankungen | NAION, LHON, toxische/metabolische Optikusneuropathie |
Zusätzliche Untersuchungen bei atypischen Fällen: Anti-AQP4-Antikörper (zum Ausschluss einer NMO), Anti-MOG-Antikörper (zum Ausschluss einer MOGAD), Serum-NfL-Test, Syphilis-Serologie (VDRL/RPR/FTA-ABS), ANA (bei SLE), ACE und Lysozym (bei Sarkoidose).
Die Standardbehandlung in Japan ist die Steroid-Pulstherapie mit Methylprednisolon 1.000 mg/Tag als intravenöse Infusion an 3 aufeinanderfolgenden Tagen. Nach der 3-tägigen Infusion wird keine orale Prednisolon-Therapie (Nachbehandlung) durchgeführt. Eine orale Steroidtherapie erhöht nachweislich die Rezidivrate und sollte nicht angewendet werden.
Auch ohne Behandlung beginnt bei etwa 80 % der Patienten innerhalb von 3 Wochen nach Beginn eine Besserung der Sehkraft, aber die Pulstherapie verkürzt die Besserungsdauer. Bei über 90 % der Fälle von Optikusneuritis ist eine Erholung des Sehvermögens zu erwarten.
Wenn die Steroid-Pulstherapie unwirksam ist, wird eine Blutreinigungsbehandlung (Plasmaaustausch) durchgeführt. Im Ausland wird Methylprednisolon 500–1.000 mg/Tag über 3–5 Tage eingesetzt. In der Optic Neuritis Treatment Trial (ONTT) verbesserte hochdosiertes intravenöses Methylprednisolon die Erholungszeit der Sehfunktion, des Kontrastsehens und des Farbsehens, zeigte jedoch keine Verbesserung der endgültigen Sehprognose.
Nach Besserung der Sehverschlechterung und Gesichtsfeldausfälle sollte in Zusammenarbeit mit einem Neurologen eine DMT zur Rezidivprophylaxe in Betracht gezogen werden.
Die wichtigsten DMT und ihre Wirksamkeit sind unten aufgeführt.
| Medikament | Wirkmechanismus | Verabreichungsweg | Relative Risikoreduktion |
|---|---|---|---|
| Interferon beta | Modulation der T-/B-Zellaktivität und Zytokinsekretion | Selbstinjektion | RR für Behinderungsprogression 0,71 |
| Glatirameracetat | Regulation regulatorischer T-Zellen | Selbstinjektion | RR für Schübe 0,82 |
| Natalizumab | Hemmung des Einstroms von Entzündungszellen ins ZNS | Infusion | RR für Schübe 0,56 |
| Fingolimod | S1P-Rezeptor-Modulation | Oral | Neue T2-Läsionen RR 0,65 |
| Teriflunomid | Pyrimidinsynthese-Hemmung | Oral | Behinderungsprogression RR 0,76 |
| Dimethylfumarat | Reduktion von oxidativem Stress und Entzündung | Oral | Rückfall RR 0,64 |
| Alemtuzumab | Anti-CD52 monoklonaler Antikörper | Infusion | Behinderungsprogression RR 0,44 |
Anti-CD20-monoklonale Antikörper (Ocrelizumab, Rituximab, Ofatumumab) haben sich als Standardtherapie für schubförmige MS etabliert3).
Selbst ohne Hirnläsionen entwickeln 25 % der Patienten mit Optikusneuritis nach 15 Jahren eine MS, bei Vorliegen von Hirnläsionen sind es 78 %.
Selbst ohne Läsionen im MRT entwickeln 25 % der Patienten nach 15 Jahren eine MS; bei Hirnläsionen sind es 78 %. Patienten mit Optikusneuritis sollten in Zusammenarbeit mit der Neurologie hinsichtlich einer DMT zur Rezidivprophylaxe evaluiert werden.
MS gilt als Autoimmunerkrankung. T-Lymphozyten erkennen Myelin als fremd und aktivieren Makrophagen, Zytokine und Antikörper, die Myelin und Axone zerstören. Der Myelinverlust beeinträchtigt die Leitung elektrischer Impulse und stört die Nervensignalübertragung.
Aktive Plaque
Schaumzell-Makrophagen : Ansammlung von Makrophagen, die die Myelinscheide phagozytiert haben.
Perivaskuläre Manschettenbildung (perivascular cuffing) : charakteristisches Bild von Lymphozyten, die die Blutgefäße umgeben.
Ödematöse fokale demyelinisierende Läsionen : treten während akuter Schübe auf.
Chronische Plaque
Myelinverlust : nachweisbar mit Luxol-Fast-Blau-Färbung. Axone bleiben erhalten, aber die Remyelinisierung ist unvollständig.
NAWM-Läsionen : diffuse Gliose, Mikroglia-Aktivierung und BBB-Zerstörung in normal erscheinender weißer Substanz. Sie korrelieren stärker mit klinischen Beeinträchtigungen als fokale Läsionen der weißen Substanz.
Oligodendrozyten sind für die Remyelinisierung des ZNS verantwortlich1). Sie ist abhängig von adulten Oligodendrozyten-Vorläuferzellen (OPC), aber vorhandene reife Oligodendrozyten können nicht zur Remyelinisierung beitragen1).
Die Hauptursachen für das Scheitern der Remyelinisierung sind folgende1).
Darüber hinaus werden auch kortikale und subkortikale Schädigungen der grauen Substanz beobachtet, und es ist bekannt, dass die Bildung von B-Zell-Follikel-ähnlichen lymphatischen Strukturen in den Meningen zu einem schwereren klinischen Verlauf führt 1).
Dies ist ein neuartiger Ansatz, der durch Hemmung von CD40L die Kostimulation zwischen T-Zellen und antigenpräsentierenden Zellen (einschließlich B-Zellen) blockiert.
In der Phase-2-Studie von Vermersch et al. (N Engl J Med 2024) zeigte Frexalimab im Vergleich zu Placebo eine deutliche Wirksamkeit bei MRT-Ergebnissen (neue gadoliniumanreichernde Läsionen in Woche 8–12), und eine Senkung des Serum-NfL, eines Biomarkers für Nervengewebsschäden, wurde ebenfalls bestätigt 3). Für die progressive MS wird auch eine Inaktivierung von Mikroglia und Makrophagen erwartet, und die Blockade des CD40L-Signals an Mikroglia am Plaque-Rand könnte theoretisch eine Neuroprotektion ermöglichen 3).
Die Etablierung eines klinischen Vorteils gegenüber den derzeitigen hochwirksamen DMT (Anti-CD20-Medikamente) wird als zukünftige Herausforderung angesehen 3).
Es wurde gezeigt, dass Ferroptose, ein eisenabhängiger Zelltod, an der neuronalen Zerstörung bei MS beteiligt ist.
Tang et al. (2025) kommentieren die Studie von Woo et al. (Cell, 2024) und berichten über eine Kaskade: Glutamat-Exzitotoxizität → Kalziumüberladung → endoplasmatischer Retikulum-Stress → Dissoziation von STING1 von STIM1 → Aktivierung des nicht-kanonischen Signalwegs → Autophagie → autophagischer Abbau von GPX4 (Enzym zur Neutralisierung von Lipidperoxidation) → Ferroptose 4). Eine erhöhte STING1-Expression in Neuronen wurde sowohl in menschlichen MS-Proben als auch in Mausmodellen bestätigt. STING1-Inhibitoren (C176, H151) reduzierten den autophagieabhängigen GPX4-Abbau in Tiermodellen und zeigten neuroprotektive Wirkung 4).
Im Forschungsstadium gelten die Neuroprotektion durch Inaktivierung von Mikroglia und Makrophagen mittels des CD40L-Inhibitors Frexalimab 3) und die Unterdrückung der Ferroptose (Eisen-abhängiger Zelltod) durch STING1-Inhibition 4) als vielversprechend. Beide befinden sich derzeit in der klinischen Prüfung/Forschung und sind keine Standardtherapie.