SM-RR
Forma recidivante-remittente (SM-RR) : il sottotipo più comune. Le recidive durano più di 24 ore, con remissione completa o parziale tra gli attacchi.

La sclerosi multipla (SM) è una malattia infiammatoria demielinizzante della sostanza bianca del sistema nervoso centrale (SNC), caratterizzata da sintomi neurologici variabili con ricadute e remissioni. Le lesioni sclerotiche cicatriziali dovute a gliosi sono tipiche, e di solito è coinvolto solo il SNC, mentre il sistema nervoso periferico è risparmiato.
La prevalenza stimata negli Stati Uniti è di 1-1,5 per 1.000 persone1). Nel mondo, 2,1 milioni di persone sono affette, con una distribuzione maggiore nelle regioni ad alta latitudine dell’emisfero settentrionale e meridionale. L’età media di insorgenza è tra 15 e 45 anni, e l’età media alla diagnosi è di 30 anni. La fascia d’età più colpita è 15-50 anni, con predominanza femminile (picco alla fine dei 20 anni), e un rapporto maschi:femmine di 1:2,9.
La SM ha quattro sottotipi principali. La SM-RR (recidivante-remittente) esordisce solitamente tra i 25 e i 29 anni, mentre la SM-SP si manifesta più spesso tra i 40 e i 49 anni1).
SM-RR
Forma recidivante-remittente (SM-RR) : il sottotipo più comune. Le recidive durano più di 24 ore, con remissione completa o parziale tra gli attacchi.
SM-SP
Forma secondariamente progressiva (SM-SP) : evoluzione dalla SM-RR. Accumulo progressivo di disabilità anche durante le fasi di remissione.
SM-PP
Forma primariamente progressiva (SM-PP) : accumulo progressivo di disabilità fin dall’esordio, senza recidive, con progressione lenta.
CIS
Sindrome clinicamente isolata (CIS) : primo episodio clinico che può evolvere in SM. Consente un trattamento precoce.
La SM si classifica in quattro sottotipi: SM-RR (recidivante-remittente), SM-SP (secondariamente progressiva), SM-PP (primariamente progressiva) e CIS (sindrome clinicamente isolata). Il più comune è la SM-RR, caratterizzata da recidive e remissioni. La SM-SP evolve dalla SM-RR, mentre la SM-PP è progressiva fin dall’inizio.
Nel 75% dei pazienti il primo sintomo è un singolo disturbo, il 45% presenta sintomi motori/sensoriali e il 20% sintomi visivi.
Sintomi oculari
Sintomi neurologici generali
L’esacerbazione è acuta o subacuta e dura da alcuni giorni a diversi mesi. Nell’85% dei casi si ha un miglioramento o la scomparsa dei sintomi, ma nel 10-15% persistono sequele.
Spesso si manifesta come una riduzione unilaterale e dolorosa dell’acuità visiva. Il dolore orbitario è presente nel 92% dei casi e peggiora con i movimenti oculari. Si osserva anche il fenomeno di Uhthoff, in cui l’aumento della temperatura corporea (bagno, esercizio fisico) peggiora temporaneamente i sintomi.
La causa esatta della SM è sconosciuta, ma si ritiene che un meccanismo autoimmune sia coinvolto nella sua insorgenza.
I fattori genetici sono coinvolti, ma il tasso di concordanza nei gemelli monozigoti è solo del 25-30%. Sebbene siano stati identificati il polimorfismo HLA e oltre 100 loci di rischio, si ritiene che non solo la predisposizione genetica ma anche i fattori ambientali giochino un ruolo importante nell’insorgenza della malattia.
Vengono utilizzati i criteri McDonald 2017 (revisione 2024). La dimostrazione della disseminazione temporale e spaziale (DIT/DIS) delle lesioni demielinizzanti del sistema nervoso centrale è fondamentale. Nella revisione del 2024, il nervo ottico è stato aggiunto come quinta sede topografica. In Giappone esistono anche i criteri diagnostici per la sclerosi multipla del Ministero della Salute, del Lavoro e del Welfare del 2015.
Le cinque sedi topografiche della disseminazione spaziale (DIS) sono le seguenti:
Prova di disseminazione temporale (DIT): ≥2 attacchi, o presenza simultanea di lesioni con e senza enhancement alla RM, nuove lesioni T2, o bande oligoclonali nel liquor 1).
Per la diagnosi di PPMS, oltre a una progressione della disabilità ≥1 anno, sono necessari almeno 2 dei seguenti: lesioni cerebrali T2, lesioni spinali T2 (≥2), o bande oligoclonali nel liquor 1).
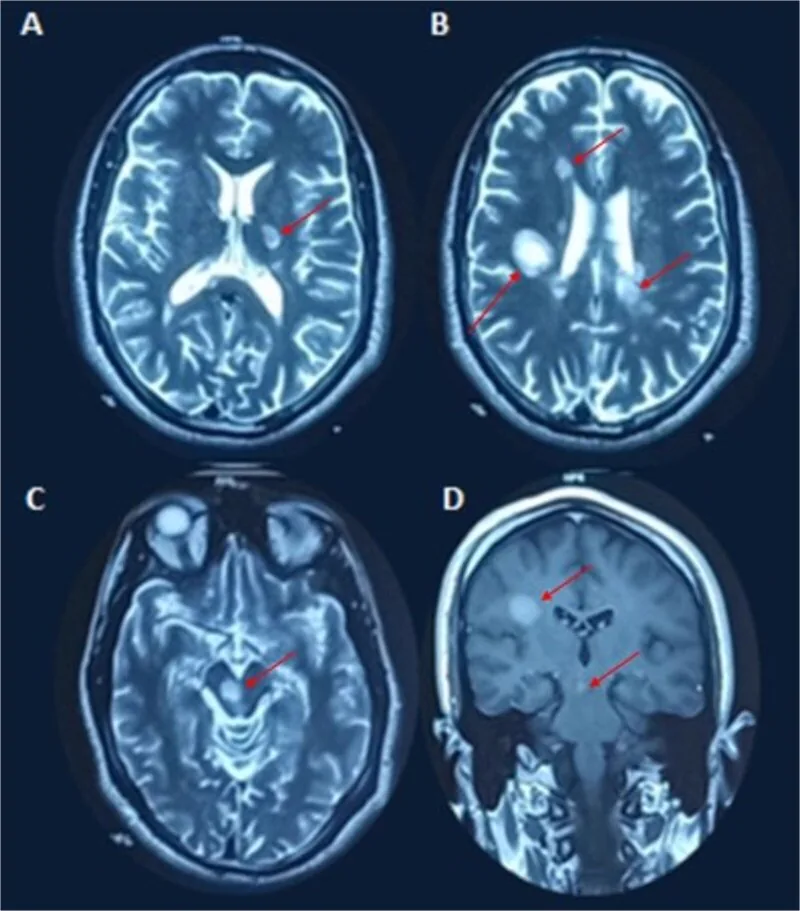
Le placche di demielinizzazione vengono rilevate come lesioni iperintense in T2 o lesioni con enhancement di gadolinio.
Utile quando la RM è inconclusiva o per prevedere la progressione della malattia 1). Può rilevare una demielinizzazione precoce e asintomatica prima della visualizzazione alla RM. Nel 65% si osserva un allungamento della latenza e una riduzione dell’ampiezza.
È importante differenziare le seguenti malattie; in casi atipici si eseguono esami aggiuntivi.
| Categoria di malattia | Principali diagnosi differenziali |
|---|---|
| Malattie demielinizzanti | NMO (malattia di Devic), ADEM, MOGAD |
| Infettivo | Sarcoidosi, tubercolosi, sifilide, malattia di Lyme |
| Autoimmune | LES, sindrome di Sjögren, malattia di Behçet |
| Malattie del nervo ottico | NAION, LHON, neuropatia ottica tossica/metabolica |
Esami aggiuntivi in casi atipici: anticorpi anti-AQP4 (per escludere NMO), anticorpi anti-MOG (per escludere MOGAD), dosaggio sierico di NfL, sierologia per sifilide (VDRL/RPR/FTA-ABS), ANA (per LES), ACE e lisozima (per sarcoidosi).
Il trattamento standard in Giappone è la terapia pulsata con metilprednisolone 1.000 mg/die per via endovenosa per 3 giorni consecutivi. Dopo l’infusione di 3 giorni, non viene somministrata terapia orale di mantenimento con prednisolone. La terapia steroidea orale aumenta il tasso di recidiva e non deve essere utilizzata.
Anche senza trattamento, circa l’80% dei pazienti inizia a migliorare l’acuità visiva entro 3 settimane dall’esordio, ma la terapia pulsata accorcia il periodo di miglioramento. In oltre il 90% dei casi di neurite ottica è prevedibile un recupero visivo.
Se la terapia pulsata con steroidi è inefficace, si esegue la plasmaferesi. All’estero si utilizza metilprednisolone 500-1.000 mg/die per 3-5 giorni. Lo studio ONTT (Optic Neuritis Treatment Trial) ha dimostrato che il metilprednisolone endovenoso ad alte dosi migliora i tempi di recupero della funzione visiva, della sensibilità al contrasto e della visione dei colori, ma non ha mostrato un miglioramento della prognosi visiva finale.
Dopo il miglioramento della riduzione dell’acuità visiva e dei difetti del campo visivo, si deve considerare una DMT per prevenire le recidive in collaborazione con un neurologo.
Le principali DMT e la loro efficacia sono elencate di seguito.
| Farmaco | Meccanismo d’azione | Via di somministrazione | Riduzione del rischio relativo |
|---|---|---|---|
| Interferone beta | Modulazione dell’attività dei linfociti T/B e della secrezione di citochine | Auto-iniezione | RR di progressione della disabilità 0,71 |
| Glatiramer acetato | Regolazione dei linfociti T regolatori | Auto-iniezione | RR di recidiva 0,82 |
| Natalizumab | Inibizione dell’infiltrazione di cellule infiammatorie nel SNC | Infusione | RR di recidiva 0,56 |
| Fingolimod | Modulazione del recettore S1P | Orale | Nuove lesioni T2 RR 0,65 |
| Teriflunomide | Inibizione della sintesi delle pirimidine | Orale | Progressione della disabilità RR 0,76 |
| Dimetilfumarato | Riduzione dello stress ossidativo e dell’infiammazione | Orale | Recidiva RR 0,64 |
| Alemtuzumab | Anticorpo monoclonale anti-CD52 | Infusione | Progressione della disabilità RR 0,44 |
Gli anticorpi monoclonali anti-CD20 (ocrelizumab, rituximab, ofatumumab) si sono affermati come terapia standard per la SM recidivante3).
Anche in assenza di lesioni cerebrali, il 25% dei pazienti con neurite ottica sviluppa SM dopo 15 anni, mentre con lesioni cerebrali la progressione verso la SM si osserva nel 78% dei casi.
Anche senza lesioni alla RM cerebrale, il 25% dei pazienti sviluppa SM dopo 15 anni; con lesioni cerebrali, il 78% progredisce verso la SM. I pazienti con neurite ottica dovrebbero essere valutati in collaborazione con la neurologia per un DMT preventivo delle recidive.
La SM è considerata una malattia autoimmune. I linfociti T riconoscono la mielina come estranea e attivano macrofagi, citochine e anticorpi che distruggono mielina e assoni. La perdita di mielina compromette la conduzione degli impulsi elettrici e altera la trasmissione dei segnali nervosi.
Placca attiva
Macrofagi schiumosi : accumulo di macrofagi che hanno fagocitato la guaina mielinica.
Infiltrazione perivascolare (perivascular cuffing) : aspetto caratteristico di linfociti che circondano i vasi sanguigni.
Lesioni demielinizzanti focali edematose : osservate durante le riacutizzazioni acute.
Placca cronica
Perdita di mielina : evidenziabile con colorazione Luxol fast blue. Gli assoni sono preservati ma la rimielinizzazione è incompleta.
Lesioni NAWM : gliosi diffusa, attivazione microgliale e rottura della BEE nella sostanza bianca apparentemente normale. Correlano maggiormente con il danno clinico rispetto alle lesioni focali della sostanza bianca.
Gli oligodendrociti sono responsabili della rimielinizzazione del SNC1). Dipende dalle cellule progenitrici degli oligodendrociti (OPC) adulte, ma gli oligodendrociti maturi esistenti non possono contribuire alla rimielinizzazione1).
Le principali cause di fallimento della rimielinizzazione sono le seguenti1).
Inoltre, si osservano anche lesioni della sostanza grigia corticale e sottocorticale, ed è noto che la formazione di strutture linfoidi follicolari a cellule B nelle meningi porta a un decorso clinico più grave 1).
Si tratta di un nuovo approccio che blocca la costimolazione tra cellule T e cellule presentanti l’antigene (incluse le cellule B) inibendo il CD40L.
Nello studio di fase 2 di Vermersch et al. (N Engl J Med 2024), frexalimab ha mostrato una chiara efficacia rispetto al placebo sugli esiti MRI (nuove lesioni captanti gadolinio a 8-12 settimane), ed è stata confermata anche una riduzione del NfL sierico, biomarcatore del danno al tessuto nervoso 3). Per la SM progressiva, ci si aspetta anche un effetto di inattivazione di microglia e macrofagi, e il blocco del segnale CD40L sulla microglia al bordo della placca potrebbe teoricamente consentire la neuroprotezione 3).
Stabilire un vantaggio clinico rispetto agli attuali DMT ad alta efficacia (farmaci anti-CD20) è considerata una sfida futura 3).
È stato dimostrato che la ferroptosi, una morte cellulare ferro-dipendente, è coinvolta nella morte neuronale nella SM.
Tang et al. (2025) commentano lo studio di Woo et al. (Cell, 2024) e riportano una cascata: eccitotossicità del glutammato → sovraccarico di calcio → stress del reticolo endoplasmatico → dissociazione di STING1 da STIM1 → attivazione della via non canonica → autofagia → degradazione autofagica di GPX4 (enzima che neutralizza la perossidazione lipidica) → ferroptosi 4). Un aumento dell’espressione di STING1 nei neuroni è stato confermato sia in campioni umani di SM che in modelli murini. Gli inibitori di STING1 (C176, H151) hanno ridotto la degradazione autofagica di GPX4 in modelli animali e hanno mostrato un effetto neuroprotettivo 4).
In fase di ricerca, la neuroprotezione attraverso l’inattivazione di microglia e macrofagi mediante l’inibitore di CD40L frexalimab 3) e la soppressione della ferroptosi (morte cellulare ferro-dipendente) mediante l’inibizione di STING1 4) sono considerate promettenti. Entrambi sono attualmente in fase di sperimentazione/ricerca e non costituiscono un trattamento standard.