RRMS
Relapsing-Remitting MS: The most common subtype. Relapses last more than 24 hours, with complete or partial remission between attacks.

Multiple sclerosis (MS) is a disease in which inflammatory demyelinating lesions occur in the white matter of the central nervous system (CNS), causing various neurological symptoms that relapse and remit. It is characterized by sclerotic lesions due to gliosis, and typically only the CNS is affected, with the peripheral nervous system spared.
The estimated prevalence in the United States is 1 to 1.5 per 1,000 people 1). Worldwide, 2.1 million people are affected, with higher distribution in high-latitude regions of the Northern and Southern Hemispheres. The average age of onset is 15 to 45 years, and the mean age at diagnosis is 30 years. The peak age of onset is 15 to 50 years, with a female predominance (peak in late 20s) and a female-to-male ratio of 1:2.9.
MS has four main subtypes. RRMS (relapsing-remitting) often begins at ages 25–29, and SPMS often begins at ages 40–491).
RRMS
Relapsing-Remitting MS: The most common subtype. Relapses last more than 24 hours, with complete or partial remission between attacks.
SPMS
Secondary Progressive MS: Transition from RRMS. Disability accumulates progressively even during remission.
PPMS
Primary Progressive MS: Disability accumulates progressively from onset. Slowly progressive without relapses.
CIS
Clinically Isolated Syndrome: The first clinical episode that may lead to MS. Enables early treatment initiation.
MS is classified into four subtypes: RRMS (relapsing-remitting), SPMS (secondary progressive), PPMS (primary progressive), and CIS (clinically isolated syndrome). The most common is RRMS, characterized by relapses and remissions. SPMS transitions from RRMS, and PPMS is progressive from onset.
In 75% of patients, the initial symptom is a single complaint; 45% present with motor/sensory symptoms, and 20% with visual symptoms.
Ocular symptoms
General neurological symptoms
Exacerbations develop acutely to subacutely and persist for days to months. Symptoms improve or resolve in 85% of cases, but sequelae remain in 10–15%.
It often presents as unilateral painful vision loss. Orbital pain is present in 92% of cases and is characteristically worsened by eye movement. Additionally, Uhthoff phenomenon, a temporary worsening of symptoms with increased body temperature (e.g., bathing, exercise), may occur.
The exact cause of MS is unknown, but autoimmune mechanisms are thought to be involved in its onset.
Genetic factors are involved, but the concordance rate in monozygotic twins is only 25-30%. Although HLA polymorphisms and over 100 risk loci have been identified, it is thought that not only genetic predisposition but also environmental factors play an important role in the onset of the disease.
The 2017 McDonald criteria (2024 revision) are used. The basic principle is to demonstrate dissemination in time and space (DIT/DIS) of demyelinating lesions in the central nervous system. In the 2024 revision, the optic nerve was added as a fifth topographic region. In Japan, the 2015 Ministry of Health, Labour and Welfare diagnostic criteria for multiple sclerosis also exist.
The five topographic regions for dissemination in space (DIS) are as follows.
Demonstration of dissemination in time (DIT): two or more attacks, or simultaneous presence of enhancing and non-enhancing lesions on MRI, new T2 lesions, or CSF oligoclonal bands can be used as alternatives 1).
For the diagnosis of PPMS, in addition to disability progression for at least one year, findings from at least two of the following are required: brain T2 lesions, spinal cord T2 lesions (two or more), or CSF oligoclonal bands 1).
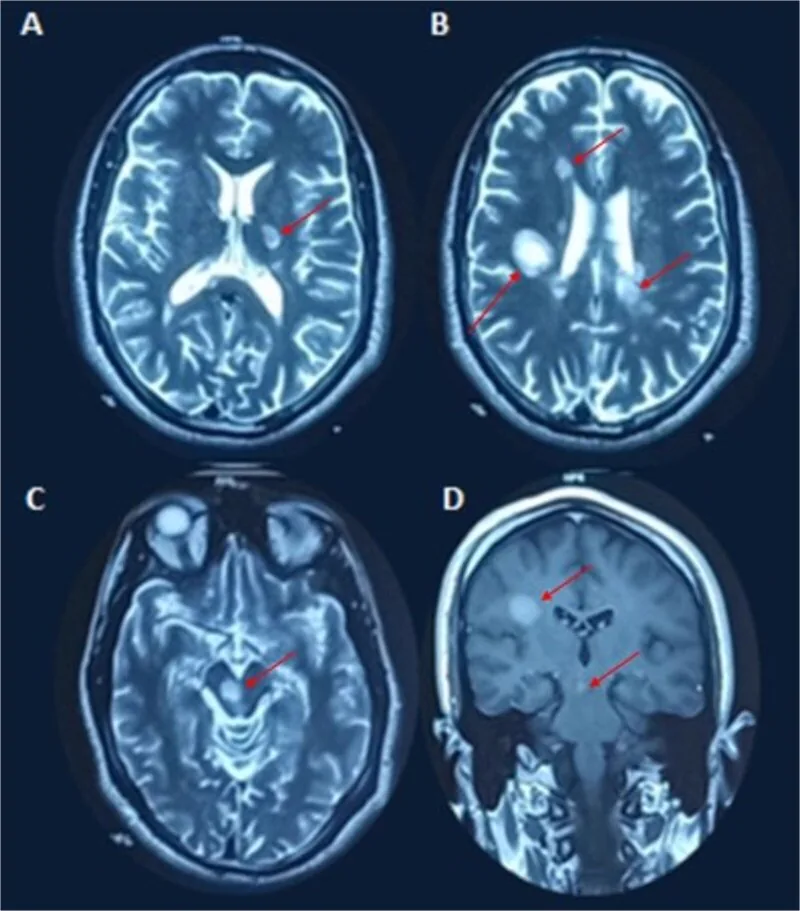
Demyelinating plaques are detected as T2 hyperintense lesions or gadolinium-enhancing lesions.
VEP is useful when MRI is inconclusive or for predicting disease progression 1). It can detect early, asymptomatic demyelination before MRI visualization. Prolonged latency and reduced amplitude are observed in 65% of cases.
Differentiation from the following diseases is important, and additional tests should be performed in atypical cases.
| Disease Category | Main Differential Diagnoses |
|---|---|
| Demyelinating Diseases | NMO (Devic’s disease), ADEM, MOGAD |
| Infectious | Sarcoidosis, tuberculosis, syphilis, Lyme disease |
| Autoimmune | SLE, Sjögren’s syndrome, Behçet’s disease |
| Optic nerve disease | NAION, LHON, toxic/metabolic optic neuropathy |
Additional tests in atypical cases: anti-AQP4 antibody (to rule out NMO), anti-MOG antibody (to rule out MOGAD), serum NfL test, syphilis serology (VDRL/RPR/FTA-ABS), ANA (SLE), ACE/lysozyme (sarcoidosis).
The standard treatment in Japan is steroid pulse therapy with intravenous methylprednisolone 1,000 mg/day for 3 consecutive days. Oral prednisolone (maintenance therapy) after the 3-day infusion is not performed. Oral steroid therapy is thought to increase the relapse rate and should not be performed.
Even without treatment, visual improvement begins within 3 weeks of onset in about 80% of cases, but pulse therapy shortens the recovery period. Visual recovery can be expected in over 90% of optic neuritis cases.
If steroid pulse therapy is ineffective, blood purification therapy (plasma exchange) is performed. Overseas, methylprednisolone 500–1,000 mg/day for 3–5 days is used. The Optic Neuritis Treatment Trial (ONTT) showed that high-dose intravenous methylprednisolone improved recovery time for visual function, contrast sensitivity, and color vision, but did not show improvement in final visual prognosis.
After improvement of visual acuity and visual field defects, DMT should be considered in collaboration with a neurologist to prevent relapse.
Major DMTs and their efficacy are shown below.
| Drug | Mechanism of action | Administration | Relative risk reduction |
|---|---|---|---|
| Interferon beta | Modulation of T/B cell activity and cytokine secretion | Self-injection | Disability progression RR 0.71 |
| Glatiramer acetate | Regulation of regulatory T cells | Self-injection | Relapse RR 0.82 |
| Natalizumab | Inhibition of inflammatory cell entry into CNS | Intravenous infusion | Relapse RR 0.56 |
| Fingolimod | S1P receptor modulator | Oral | New T2 lesions RR 0.65 |
| Teriflunomide | Pyrimidine synthesis inhibitor | Oral | Disability progression RR 0.76 |
| Dimethyl fumarate | Reduces oxidative stress and inflammation | Oral | Relapse RR 0.64 |
| Alemtuzumab | Anti-CD52 monoclonal antibody | Intravenous | Disability progression RR 0.44 |
Anti-CD20 monoclonal antibodies (ocrelizumab, rituximab, ofatumumab) have become established as standard therapy for relapsing MS 3).
Even in optic neuritis without brain lesions, MS develops in 25% after 15 years, and in cases with brain lesions, transition to MS is seen in 78%.
Even without brain MRI lesions, MS develops in 25% after 15 years; with brain lesions, transition to MS occurs in 78%. Patients who develop optic neuritis should be evaluated for DMT to prevent relapse in collaboration with neurology.
MS is considered an autoimmune disease. T lymphocytes recognize myelin as foreign and activate macrophages, cytokines, and antibodies to destroy myelin and axons. Loss of myelin impairs electrical impulse conduction and disrupts neural signal transmission.
Active Plaque
Foamy macrophages: Accumulation of macrophages that have phagocytosed myelin.
Perivascular cuffing: Characteristic finding of lymphocytes surrounding blood vessels.
Edematous focal demyelinating lesions: Seen during acute exacerbations.
Chronic Plaque
Myelin loss: Can be confirmed with Luxol fast blue staining. Axons are preserved but remyelination is incomplete.
NAWM lesions: Diffuse gliosis, microglial activation, and BBB disruption in normal-appearing white matter. They show a higher correlation with clinical disability than focal white matter lesions.
Oligodendrocytes are responsible for remyelination in the CNS1). It depends on adult oligodendrocyte precursor cells (OPCs), but existing mature oligodendrocytes cannot contribute to remyelination1).
The main causes of remyelination failure are as follows1).
Additionally, gray matter damage in the cortex and subcortical regions is observed, and when B-cell follicle-like lymphoid structures form in the meninges, it is known to lead to a more severe clinical course1).
By inhibiting CD40L, this novel approach blocks co-stimulation between T cells and antigen-presenting cells (including B cells).
In a phase 2 trial by Vermersch et al. (N Engl J Med 2024), frexalimab showed clear efficacy over placebo in MRI outcomes (new gadolinium-enhancing lesions at 8–12 weeks), and a reduction in serum NfL, a biomarker of neuronal tissue damage, was also confirmed3). For progressive MS, it is also expected to inactivate microglia and macrophages, and neuroprotection via blockade of CD40L signaling in microglia at the plaque edge is theoretically possible3).
Establishing clinical superiority over current high-efficacy DMTs (anti-CD20 drugs) remains a future challenge3).
Ferroptosis, an iron-dependent cell death, has been shown to be involved in neuronal cell death in MS.
Tang et al. (2025) reviewed the study by Woo et al. (Cell, 2024) and reported a cascade: glutamate excitotoxicity → calcium overload → endoplasmic reticulum stress → STING1 dissociates from STIM1 → atypical pathway activation → autophagy → autophagic degradation of GPX4 (an enzyme that neutralizes lipid peroxidation) → ferroptosis 4). Increased STING1 expression in neurons was confirmed in both human MS specimens and mouse models. STING1 inhibitors (C176, H151) reduced autophagy-dependent GPX4 degradation in animal models and showed neuroprotective effects 4).
At the research stage, neuroprotection through inactivation of microglia and macrophages by the CD40L inhibitor frexalimab 3) and suppression of ferroptosis (iron-dependent cell death) by STING1 inhibition 4) are considered promising. Both are currently in clinical trial or research stages and are not standard treatments.