RRMS
復發緩解型MS:最常見的亞型。復發持續超過24小時,發作間期有完全或部分緩解。

多發性硬化症(Multiple Sclerosis; MS)是一種中樞神經系統(CNS)白質出現炎性脫髓鞘病變,導致多種神經症狀反覆發作和緩解的疾病。其特徵是膠質增生導致的瘢痕硬化性病變,通常僅累及中樞神經,周圍神經系統不受影響。
美國估計患病率為1~1.5/1000人1)。全球有210萬人患病,在北半球和南半球的高緯度地區分佈較多。平均發病年齡為15~45歲,診斷時平均年齡為30歲。好發年齡為15~50歲,女性多見(20歲後期為高峰),男女比例為1:2.9。
MS有以下四種主要亞型。RRMS(復發緩解型)常在25-29歲發病,SPMS常在40-49歲發病1)。
RRMS
復發緩解型MS:最常見的亞型。復發持續超過24小時,發作間期有完全或部分緩解。
SPMS
繼發進展型MS:由RRMS轉變而來。即使在緩解期,障礙也進行性累積。
PPMS
原發進展型MS:從發病起障礙就進行性累積。無復發,緩慢進展。
CIS
臨床孤立症候群:可能發展為MS的首次臨床發作。可早期開始治療。
75%的患者初發症狀為單一主訴,45%表現為運動/感覺症狀,20%表現為視覺症狀。
眼部症狀
全身神經症狀
惡化呈急性至亞急性發作,持續數天至數月。85%的患者症狀改善或消失,但10–15%留有後遺症。
常表現為單眼疼痛性視力下降。92%出現眼眶痛,特徵為眼球運動時加重。此外,體溫升高(如洗澡、運動)導致症狀暫時惡化的Uhthoff現象也可出現。
MS的確切原因尚不清楚,但認為自體免疫機制參與發病。
遺傳因素有關,但同卵雙胞胎的一致率僅為25-30%。雖然已鑑定出HLA多型性和超過100個風險基因位點,但認為不僅遺傳易感性,環境因素也在發病中扮演重要角色。
採用2017年McDonald標準(2024年修訂版)。基本原則是證明中樞神經系統脫髓鞘病變的時間和空間多發性(DIT/DIS)。2024年修訂中,視神經被新增為第五個地形區域。在日本,也有2015年厚生勞動省的多發性硬化診斷標準。
空間多發性(DIS)的五個地形區域如下。
時間多發性(DIT)的證明:兩次或以上發作,或MRI上顯影與非顯影病灶同時存在、新T2病灶、CSF寡克隆帶可作為替代 1)。
PPMS的診斷需要至少1年的殘疾進展,並且滿足以下至少兩項:腦T2病灶、脊髓T2病灶(兩個或以上)、CSF寡克隆帶 1)。
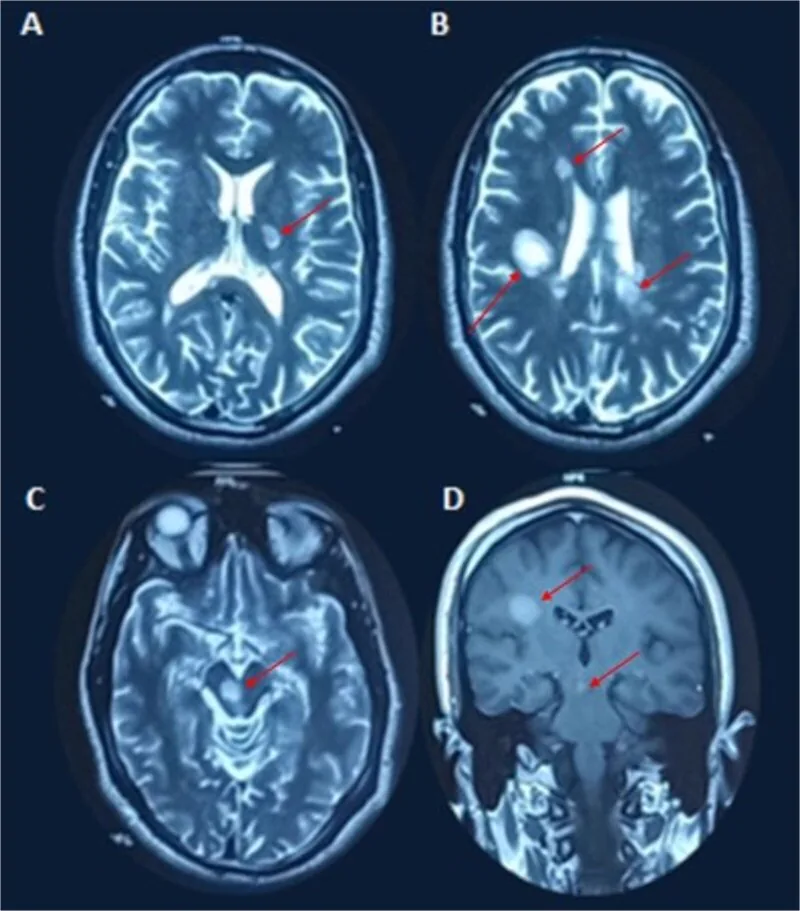
脫髓鞘斑塊表現為T2高信號病灶或釓增強病灶。
當MRI不確定或用於預測疾病進展時,VEP有用1)。它可以在MRI可視化之前檢測早期、無症狀的脫髓鞘。65%的病例出現潛伏期延長和振幅降低。
與以下疾病的鑑別很重要,非典型病例應進行附加檢查。
| 疾病類別 | 主要鑑別診斷 |
|---|---|
| 脫髓鞘疾病 | NMO(德維克病)、ADEM、MOGAD |
| 感染性 | 類肉瘤病、結核、梅毒、萊姆病 |
| 自體免疫性 | 系統性紅斑狼瘡、修格連氏症候群、貝西氏症 |
| 視神經疾病 | 非動脈炎性前部缺血性視神經病變、LHON、中毒性/代謝性視神經病變 |
非典型病例的額外檢查:抗AQP4抗體(排除NMO)、抗MOG抗體(排除MOGAD)、血清NfL檢測、梅毒血清學檢查(VDRL/RPR/FTA-ABS)、ANA(SLE)、ACE/溶菌酶(類肉瘤病)。
日本的標準治療是甲基培尼皮質醇1000毫克/日靜脈滴注,連續3天的類固醇脈衝療法。3天滴注後不進行口服培尼皮質醇(後續治療)。口服類固醇治療被認為會增加復發率,不應進行。
即使不治療,約80%的病例在發病後3週內視力開始改善,但脈衝療法可縮短恢復期。超過90%的視神經炎病例可望恢復視力。
如果類固醇脈衝療法無效,則進行血液淨化療法(血漿置換)。在國外,使用甲基培尼皮質醇500-1000毫克/日,持續3-5天。視神經炎治療試驗(ONTT)顯示,高劑量甲基培尼皮質醇靜脈注射可改善視功能、對比敏感度和色覺的恢復時間,但未顯示最終視力預後的改善。
視力下降和視野缺損改善後,應與神經內科醫師合作考慮DMT以預防復發。
主要DMT及其有效性如下所示。
| 藥物 | 作用機轉 | 給藥方式 | 相對風險降低 |
|---|---|---|---|
| 干擾素β | 調節T/B細胞活性與細胞激素分泌 | 自行注射 | 殘疾進展RR 0.71 |
| 醋酸格拉替雷 | 調節調節性T細胞 | 自行注射 | 復發RR 0.82 |
| 那他珠單抗 | 抑制發炎細胞進入中樞神經系統 | 靜脈輸注 | 復發RR 0.56 |
| 芬戈莫德 | S1P受體調節劑 | 口服 | 新T2病灶RR 0.65 |
| 特立氟胺 | 嘧啶合成抑制劑 | 口服 | 殘疾進展RR 0.76 |
| 富馬酸二甲酯 | 減輕氧化壓力和發炎 | 口服 | 復發RR 0.64 |
| 阿侖單抗 | 抗CD52單株抗體 | 靜脈輸注 | 殘疾進展RR 0.44 |
抗CD20單株抗體(ocrelizumab、rituximab、ofatumumab)已成為復發型MS的標準治療3)。
即使沒有腦部病變的視神經炎,15年後仍有25%發生MS;有腦部病變者,78%會轉化為MS。
即使腦MRI無病變,15年後仍有25%發生MS;有腦病變者,78%轉化為MS。發生視神經炎的患者應與神經內科協作,考慮使用DMT預防復發。
MS被認為是一種自體免疫疾病。T淋巴球將髓鞘視為異物,激活巨噬細胞、細胞激素和抗體,破壞髓鞘和軸突。髓鞘缺失導致電脈衝傳導障礙,神經訊號傳遞受損。
活動性斑塊
泡沫狀巨噬細胞:吞噬髓鞘的巨噬細胞聚集。
血管周圍浸潤(perivascular cuffing):淋巴細胞圍繞血管的特徵性表現。
水腫性局部脫髓鞘病變:見於急性惡化期。
慢性斑塊
髓鞘脫失:可用Luxol fast blue染色確認。軸突保留但再髓鞘化不完全。
NAWM病變:外觀正常的白質中瀰漫性神經膠質增生、微膠質細胞激活和血腦屏障破壞。與局部白質病變相比,與臨床失能的相关性更高。
寡突膠細胞負責中樞神經系統的再髓鞘化1)。這依賴於成體寡突膠細胞前驅細胞(OPC),但現有的成熟寡突膠細胞不能參與再髓鞘化1)。
再髓鞘化失敗的主要原因如下1)。
此外,也觀察到皮質與皮質下灰質損傷,當腦膜中形成B細胞濾泡樣淋巴結構時,已知會導致更嚴重的臨床病程1)。
透過抑制CD40L,阻斷T細胞與抗原呈現細胞(包括B細胞)共刺激的新方法。
Vermersch等人(N Engl J Med 2024)的第二期試驗顯示,frexalimab在MRI結果(8-12週的新釓增強病灶)方面顯示出優於安慰劑的明確療效,並且血清NfL(神經組織損傷的生物標誌物)的降低也得到證實3)。對於進展型MS,也預期具有使微膠質細胞和巨噬細胞失活的效果,理論上可透過阻斷斑塊邊緣微膠質細胞的CD40L訊號實現神經保護3)。
目前面臨的挑戰是確立其相對於現有高效DMT(抗CD20藥物)的臨床優勢3)。
鐵依賴性細胞死亡——鐵死亡已被證明參與MS的神經元死亡。
Tang等人(2025)解讀了Woo等人(Cell, 2024)的研究,報告了以下級聯反應:穀氨酸興奮毒性→鈣超載→內質網應激→STING1從STIM1解離→非典型通路激活→自噬→GPX4(脂質過氧化中和酶)的自噬降解→鐵死亡4)。在人類多發性硬化症標本和小鼠模型中均證實了神經元中STING1表達升高。STING1抑制劑(C176、H151)在動物模型中減少了自噬依賴性GPX4降解,並顯示出神經保護作用4)。
在研究階段,通過CD40L抑制劑frexalimab使微膠質細胞和巨噬細胞失活從而實現神經保護3),以及通過抑制STING1來抑制鐵死亡(鐵依賴性細胞死亡)4)被認為是有前景的。兩者目前均處於臨床試驗或研究階段,並非標準治療。