RRMS
EM recurrente-remitente: El subtipo más común. Las recaídas duran más de 24 horas, con remisión completa o parcial entre los ataques.

La esclerosis múltiple (EM) es una enfermedad en la que se producen lesiones desmielinizantes inflamatorias en la sustancia blanca del sistema nervioso central (SNC), causando diversos síntomas neurológicos que recaen y remiten. Se caracteriza por lesiones esclerosas debidas a gliosis y, típicamente, solo el SNC se ve afectado, mientras que el sistema nervioso periférico no se daña.
La prevalencia estimada en Estados Unidos es de 1 a 1.5 por cada 1,000 personas 1). En todo el mundo, 2.1 millones de personas están afectadas, con una mayor distribución en regiones de alta latitud de los hemisferios norte y sur. La edad promedio de inicio es de 15 a 45 años, y la edad media al diagnóstico es de 30 años. La edad de mayor incidencia es de 15 a 50 años, con predominio femenino (pico a finales de los 20 años) y una proporción mujer:hombre de 1:2.9.
La EM tiene cuatro subtipos principales. La RRMS (recurrente-remitente) suele comenzar entre los 25 y 29 años, y la SPMS entre los 40 y 49 años1).
RRMS
EM recurrente-remitente: El subtipo más común. Las recaídas duran más de 24 horas, con remisión completa o parcial entre los ataques.
SPMS
EM secundaria progresiva: Transición desde RRMS. La discapacidad se acumula progresivamente incluso durante la remisión.
PPMS
EM primaria progresiva: La discapacidad se acumula progresivamente desde el inicio. Progresión lenta sin recaídas.
CIS
Síndrome clínicamente aislado: El primer episodio clínico que puede convertirse en EM. Permite iniciar el tratamiento temprano.
La EM se clasifica en cuatro subtipos: RRMS (recurrente-remitente), SPMS (secundaria progresiva), PPMS (primaria progresiva) y CIS (síndrome clínicamente aislado). El más común es RRMS, caracterizado por recaídas y remisiones. SPMS es una transición desde RRMS, y PPMS es progresiva desde el inicio.
En el 75% de los pacientes, el síntoma inicial es una queja única; el 45% presenta síntomas motores/sensoriales y el 20% síntomas visuales.
Síntomas oculares
Síntomas neurológicos generales
Las exacerbaciones se desarrollan de forma aguda a subaguda y persisten durante días a meses. Los síntomas mejoran o se resuelven en el 85% de los casos, pero persisten secuelas en el 10–15%.
A menudo se presenta como pérdida visual unilateral dolorosa. El dolor orbitario está presente en el 92% de los casos y empeora característicamente con el movimiento ocular. Además, puede ocurrir el fenómeno de Uhthoff, un empeoramiento temporal de los síntomas con el aumento de la temperatura corporal (p. ej., baño, ejercicio).
Se desconoce la causa exacta de la EM, pero se cree que los mecanismos autoinmunitarios están involucrados en su aparición.
Los factores genéticos están involucrados, pero la tasa de concordancia en gemelos monocigóticos es solo del 25-30%. Aunque se han identificado polimorfismos de HLA y más de 100 loci de riesgo, se cree que no solo la predisposición genética sino también los factores ambientales juegan un papel importante en la aparición de la enfermedad.
Se utilizan los criterios McDonald de 2017 (revisión de 2024). El principio básico es demostrar la diseminación en tiempo y espacio (DIT/DIS) de las lesiones desmielinizantes del sistema nervioso central. En la revisión de 2024, el nervio óptico se agregó como una quinta región topográfica. En Japón, también existen los criterios diagnósticos de esclerosis múltiple del Ministerio de Salud, Trabajo y Bienestar de 2015.
Las cinco regiones topográficas para la diseminación en espacio (DIS) son las siguientes.
Demostración de diseminación en el tiempo (DIT): dos o más ataques, o presencia simultánea de lesiones con y sin realce en RM, nuevas lesiones T2, o bandas oligoclonales en LCR pueden usarse como alternativa 1).
Para el diagnóstico de PPMS, además de la progresión de la discapacidad durante al menos un año, se requieren hallazgos de al menos dos de los siguientes: lesiones T2 cerebrales, lesiones T2 en médula espinal (dos o más), o bandas oligoclonales en LCR 1).
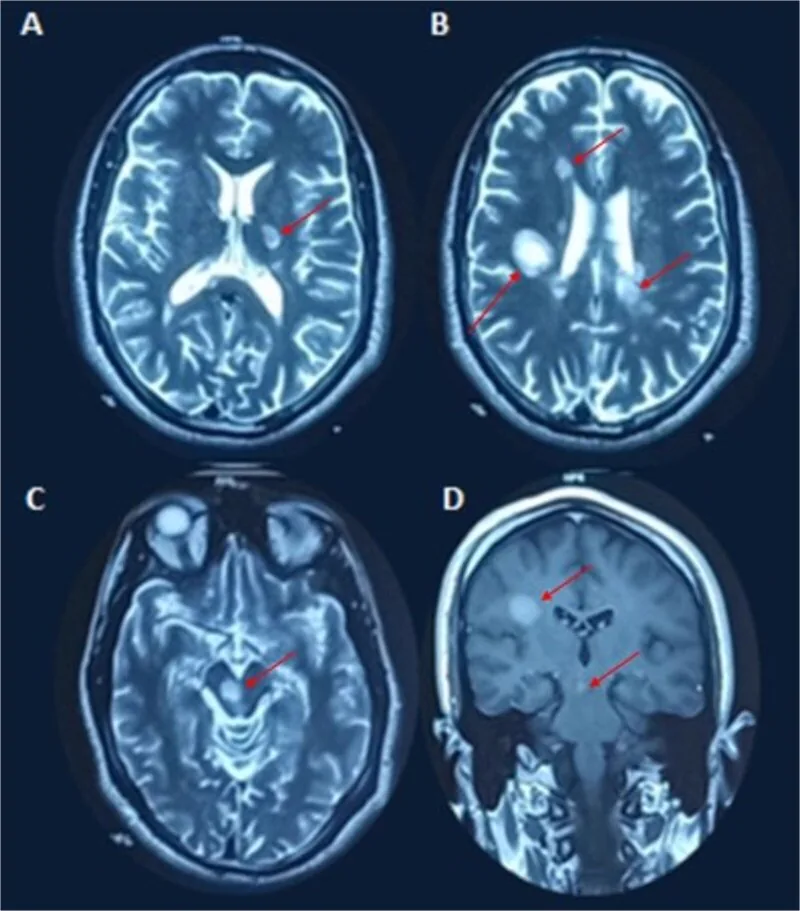
Las placas desmielinizantes se detectan como lesiones hiperintensas en T2 o lesiones con realce de gadolinio.
El VEP es útil cuando la resonancia magnética no es concluyente o para predecir la progresión de la enfermedad 1). Puede detectar desmielinización temprana y asintomática antes de la visualización por resonancia magnética. Se observa latencia prolongada y amplitud reducida en el 65% de los casos.
La diferenciación de las siguientes enfermedades es importante, y se deben realizar pruebas adicionales en casos atípicos.
| Categoría de Enfermedad | Principales Diagnósticos Diferenciales |
|---|---|
| Enfermedades Desmielinizantes | NMO (enfermedad de Devic), ADEM, MOGAD |
| Infeccioso | Sarcoidosis, tuberculosis, sífilis, enfermedad de Lyme |
| Autoinmune | LES, síndrome de Sjögren, enfermedad de Behçet |
| Enfermedad del nervio óptico | NAION, LHON, neuropatía óptica tóxica/metabólica |
Pruebas adicionales en casos atípicos: anticuerpos anti-AQP4 (para descartar NMO), anticuerpos anti-MOG (para descartar MOGAD), prueba de NfL sérico, serología de sífilis (VDRL/RPR/FTA-ABS), ANA (LES), ECA/lisozima (sarcoidosis).
El tratamiento estándar en Japón es la terapia de pulso con esteroides con metilprednisolona intravenosa 1,000 mg/día durante 3 días consecutivos. No se realiza prednisolona oral (terapia de mantenimiento) después de la infusión de 3 días. Se cree que la terapia con esteroides orales aumenta la tasa de recaídas y no debe realizarse.
Incluso sin tratamiento, la mejoría visual comienza dentro de las 3 semanas del inicio en aproximadamente el 80% de los casos, pero la terapia de pulso acorta el período de recuperación. Se puede esperar recuperación visual en más del 90% de los casos de neuritis óptica.
Si la terapia de pulso con esteroides no es efectiva, se realiza terapia de purificación sanguínea (plasmaféresis). En el extranjero, se utiliza metilprednisolona 500–1,000 mg/día durante 3–5 días. El Ensayo de Tratamiento de Neuritis Óptica (ONTT) mostró que la metilprednisolona intravenosa en dosis altas mejoró el tiempo de recuperación de la función visual, la sensibilidad al contraste y la visión del color, pero no demostró mejoría en el pronóstico visual final.
Después de la mejoría de la agudeza visual y los defectos del campo visual, se debe considerar la DMT en colaboración con un neurólogo para prevenir recaídas.
Los principales DMT y su eficacia se muestran a continuación.
| Fármaco | Mecanismo de acción | Vía de administración | Reducción del riesgo relativo |
|---|---|---|---|
| Interferón beta | Modulación de la actividad de células T/B y secreción de citocinas | Autoinyección | RR de progresión de discapacidad 0.71 |
| Acetato de glatiramer | Regulación de células T reguladoras | Autoinyección | RR de recaída 0.82 |
| Natalizumab | Inhibición de la entrada de células inflamatorias al SNC | Infusión intravenosa | RR de recaída 0.56 |
| Fingolimod | Modulador del receptor S1P | Oral | Nuevas lesiones T2 RR 0.65 |
| Teriflunomida | Inhibidor de la síntesis de pirimidina | Oral | Progresión de discapacidad RR 0.76 |
| Dimetilfumarato | Reduce el estrés oxidativo y la inflamación | Oral | Recaída RR 0.64 |
| Alemtuzumab | Anticuerpo monoclonal anti-CD52 | Intravenoso | Progresión de discapacidad RR 0.44 |
Los anticuerpos monoclonales anti-CD20 (ocrelizumab, rituximab, ofatumumab) se han establecido como tratamiento estándar para la EM recurrente 3).
Incluso en la neuritis óptica sin lesiones cerebrales, el 25% desarrolla EM después de 15 años, y en casos con lesiones cerebrales, se observa transición a EM en el 78%.
Incluso sin lesiones en la RM cerebral, el 25% desarrolla EM después de 15 años; con lesiones cerebrales, la transición a EM ocurre en el 78%. Los pacientes que desarrollan neuritis óptica deben ser evaluados para DMT para prevenir recaídas en colaboración con neurología.
La EM se considera una enfermedad autoinmune. Los linfocitos T reconocen la mielina como extraña y activan macrófagos, citocinas y anticuerpos para destruir la mielina y los axones. La pérdida de mielina altera la conducción de impulsos eléctricos y perjudica la transmisión de señales nerviosas.
Placa activa
Macrófagos espumosos: Acumulación de macrófagos que han fagocitado mielina.
Infiltrado perivascular (perivascular cuffing): Hallazgo característico de linfocitos rodeando los vasos sanguíneos.
Lesiones desmielinizantes focales edematosas: Se observan durante las exacerbaciones agudas.
Placa crónica
Pérdida de mielina: Se puede confirmar con la tinción de Luxol fast blue. Los axones se conservan pero la remielinización es incompleta.
Lesiones NAWM: Gliosis difusa, activación de microglía y ruptura de la BHE en la sustancia blanca de apariencia normal. Muestran una correlación más alta con la discapacidad clínica que las lesiones focales de sustancia blanca.
Los oligodendrocitos son responsables de la remielinización en el SNC1). Depende de las células precursoras de oligodendrocitos (OPC) adultas, pero los oligodendrocitos maduros existentes no pueden contribuir a la remielinización1).
Las principales causas del fracaso de la remielinización son las siguientes1).
Además, también se observa daño en la sustancia gris cortical y subcortical, y se sabe que cuando se forman estructuras linfoides similares a folículos de células B en las meninges, se produce un curso clínico más grave1).
Al inhibir CD40L, este nuevo enfoque bloquea la coestimulación entre las células T y las células presentadoras de antígenos (incluidas las células B).
En un ensayo de fase 2 de Vermersch et al. (N Engl J Med 2024), frexalimab mostró una eficacia clara frente a placebo en los resultados de resonancia magnética (nuevas lesiones con realce de gadolinio a las 8-12 semanas), y también se confirmó una reducción del NfL sérico, un biomarcador de daño del tejido nervioso3). Para la EM progresiva, también se espera un efecto de inactivación de microglía y macrófagos, y teóricamente es posible la neuroprotección mediante el bloqueo de la señalización de CD40L en la microglía en el borde de la placa3).
Establecer la superioridad clínica sobre los DMT de alta eficacia actuales (fármacos anti-CD20) sigue siendo un desafío futuro3).
Se ha demostrado que la ferroptosis, una muerte celular dependiente de hierro, está implicada en la muerte neuronal en la EM.
Tang et al. (2025) revisaron el estudio de Woo et al. (Cell, 2024) e informaron una cascada: excitotoxicidad por glutamato → sobrecarga de calcio → estrés del retículo endoplásmico → disociación de STING1 de STIM1 → activación de la vía atípica → autofagia → degradación autofágica de GPX4 (enzima que neutraliza la peroxidación lipídica) → ferroptosis 4). Se confirmó un aumento de la expresión de STING1 en neuronas tanto en muestras humanas de EM como en modelos de ratón. Los inhibidores de STING1 (C176, H151) redujeron la degradación autofágica de GPX4 en modelos animales y mostraron efectos neuroprotectores 4).
En la etapa de investigación, la neuroprotección mediante la inactivación de microglía y macrófagos por el inhibidor de CD40L frexalimab 3) y la supresión de la ferroptosis (muerte celular dependiente de hierro) mediante la inhibición de STING1 4) se consideran prometedores. Ambos se encuentran actualmente en fase de ensayo clínico o investigación y no son tratamientos estándar.