La enfermedad de Behçet es una enfermedad inflamatoria sistémica refractaria de causa desconocida que presenta cuatro síntomas principales: úlceras aftosas orales, lesiones oculares, úlceras genitales y lesiones cutáneas. La inflamación es aguda y transitoria, pero la enfermedad se caracteriza por recaídas repetidas. Ocurre en adultos jóvenes y de mediana edad (20–50 años) con una proporción hombre:mujer de aproximadamente 1:1. La uveítis ocurre en aproximadamente el 70% de los pacientes masculinos y el 45% de las pacientes femeninas, con casos graves más frecuentes en hombres jóvenes.
Se observa con frecuencia a lo largo de la Ruta de la Seda, desde el Mediterráneo hasta China y Japón. Aproximadamente el 50% de los pacientes son positivos para HLA-B51 (15% en la población general)5), y también se ha informado que HLA-A26 (A*2601) es un alelo de riesgo independiente. Se cree que la predisposición inmunogenética y los factores ambientales están involucrados en el inicio. Esta enfermedad alguna vez tuvo una alta tasa de ceguera, pero los avances en inmunosupresores y agentes biológicos han reducido la tasa de ceguera.
La enfermedad de Behçet está registrada como enfermedad intratable designada (No. 56)8).
Características de la enfermedad
Edad de inicio: Adultos jóvenes y de mediana edad (20–50 años)
Diferencia de sexo: Proporción hombre:mujer aproximadamente 1:1. Las lesiones oculares suelen ser graves en hombres jóvenes.
Distribución geográfica: Alta frecuencia a lo largo de la Ruta de la Seda (Mediterráneo, Medio Oriente, Asia Oriental).
Antecedentes genéticos: HLA-B51 positivo (aproximadamente el 50% de los pacientes), HLA-A26 también es un factor de riesgo independiente 5).
4 Síntomas Principales
Úlceras aftosas orales: Úlceras recurrentes dolorosas. A menudo son el síntoma inicial.
La proporción de la enfermedad de Behçet entre todas las uveítis ha ido disminuyendo con el tiempo. En la encuesta epidemiológica de 2002 fue del 6,2% (tercer lugar), pero en 2009 disminuyó al 3,9% (sexto lugar) 5). Además de una disminución en el número de pacientes, también se ha informado una evolución más leve de la enfermedad 4).
Q¿Qué tipo de enfermedad es la enfermedad de Behçet?
A
La enfermedad de Behçet es una enfermedad inflamatoria sistémica que se repite, caracterizada por cuatro síntomas principales: úlceras aftosas orales, síntomas oculares, úlceras genitales y síntomas cutáneos. Se desconoce la causa, pero se sospecha un mecanismo autoinmune. Es más común en regiones a lo largo de la Ruta de la Seda, y se cree que están involucrados antecedentes genéticos (HLA-B51) y factores ambientales. Las lesiones oculares son particularmente importantes y pueden provocar ceguera sin el tratamiento adecuado.
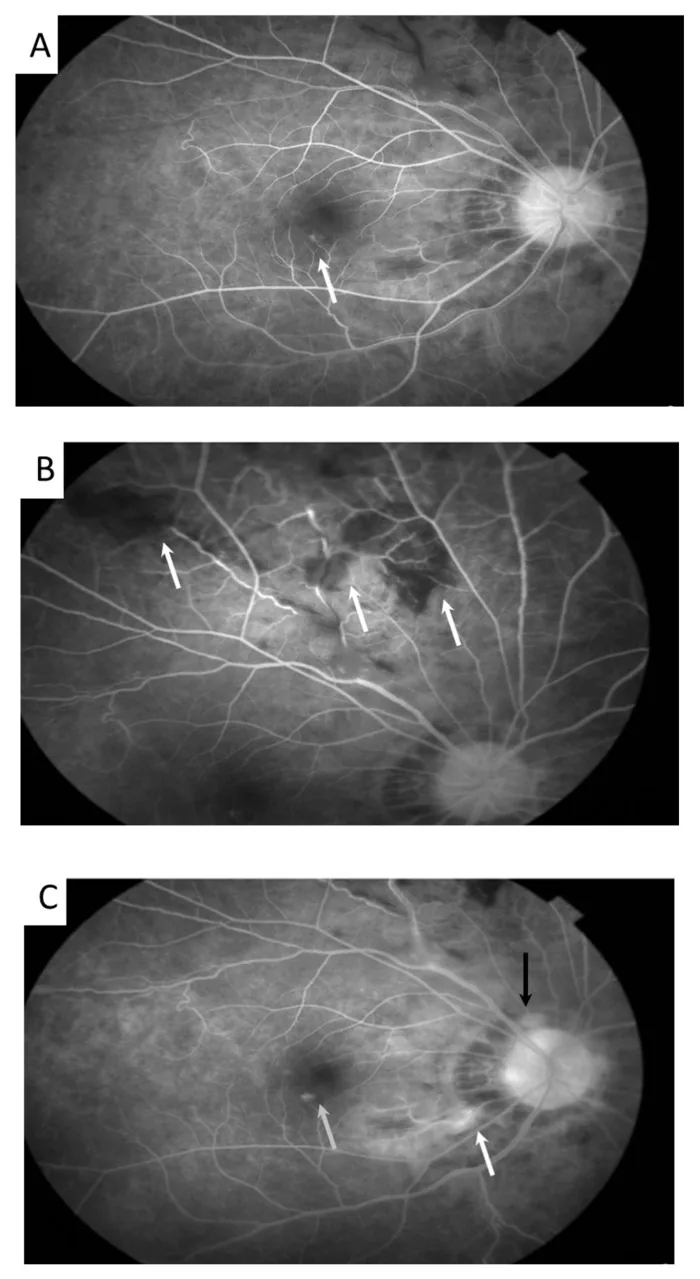
Meng PP, et al. Use of Ultra-Widefield Fluorescein Angiography to Guide the Treatment to Idiopathic Retinal Vasculitis, Aneurysms, and Neuroretinitis-Case Report and Literature Review. Medicina (Kaunas). 2022. Figure 2. PMCID: PMC9611749. License: CC BY.
En la imagen de autofluorescencia, se observan dilataciones aneurismáticas de las arteriolas retinianas (flecha) y tortuosidad venosa. Esto corresponde a los hallazgos de vasculitis retiniana discutidos en la sección “2. Síntomas Principales y Hallazgos Clínicos”.
Los síntomas subjetivos oculares incluyen visión borrosa, disminución de la agudeza visual y miodesopsias durante los ataques inflamatorios. La inflamación a menudo recurre en un ojo en diferentes momentos, volviéndose bilateral en aproximadamente el 90% de los casos. En casos graves, los ataques pueden ocurrir varias veces al mes, mientras que en casos leves ocurren aproximadamente una vez al año y continúan durante varios años o más de una década. La retinitis localizada en la mácula puede recurrir, causando pérdida irreversible de la visión.
Los síntomas sistémicos incluyen principalmente úlceras aftosas orales (aproximadamente 90%), lesiones cutáneas (aproximadamente 75%) y úlceras genitales (aproximadamente 50%). Las úlceras aftosas orales ocurren comúnmente en la lengua, mucosa bucal, labios y encías, y son úlceras dolorosas con enrojecimiento circundante. Son casi siempre presentes en esta enfermedad y a menudo son el síntoma inicial. Sanan sin cicatriz en 10 días, pero recurren repetidamente.
Los síntomas secundarios incluyen artritis sin deformidad ni rigidez, epididimitis, enfermedad de Behçet gastrointestinal (úlceras ileocecales), enfermedad de Behçet vascular (vasculitis) y enfermedad de Behçet neurológica (meningoencefalitis). Dado que los síntomas extraoculares y los hallazgos oculares no siempre están vinculados, es importante evaluar el estado sistémico además de la observación oftalmológica3).
Los siguientes dos síntomas oculares recurren paroxísticamente.
(1) Iridociclitis aguda, a veces con hipopión.
(2) Opacidades vítreas difusas, retinitis coroidea y vasculitis retiniana.
Hallazgos en fase temprana y activa de los ataques oculares: Inflamación ocular paroxística. Precipitados queráticos finos, células inflamatorias en cámara anterior e hipopión. La exudación de fibrina en la cámara anterior suele estar ausente. No se forman nódulos iridianos ni del ángulo (no granulomatoso). En el segmento posterior, las opacidades vítreas, exudados retinianos y hemorragias se resuelven característicamente con relativa rapidez (1–2 semanas)3).
Hallazgos tardíos3): Atrofia retinoidea coroidea, vascularización retiniana blanquecina, palidez del disco óptico → que conduce a disfunción visual grave.
El hipopión en la enfermedad de Behçet es fluido debido a la infiltración predominante de neutrófilos, formando un nivel horizontal limpio. Se desplaza fácilmente con los cambios de posición corporal, diferenciándose del hipopión viscoso de la uveítis asociada a HLA-B273). Los precipitados queráticos son finos y polvorientos, mostrando hallazgos no granulomatosos.
Durante la exacerbación aguda de la uveítis retiniana, se observan opacidades vítreas, vasculitis retiniana, exudados retinianos blancos y hemorragias. Los exudados blancos se deben a la infiltración de leucocitos y la inflamación isquémica de las fibras nerviosas, y característicamente se resuelven con relativa rapidez (en aproximadamente una semana). La vasculitis oclusiva puede causar hemorragias que se asemejan a la oclusión de la rama venosa retiniana.
La angiografía con fluoresceína a menudo revela una fuga extensa y vigorosa de los capilares retinianos (fuga en forma de helecho) incluso cuando no hay ataque ocular, lo que se considera característico de esta enfermedad.
BOS24 (Puntuación de ataque ocular de la enfermedad de Behçet 24)6)
Un sistema de puntuación que cuantifica el grado de inflamación de cada ataque ocular. Consta de seis ítems: inflamación de la cámara anterior (máx. 4 puntos), opacidad vítrea (máx. 4 puntos), lesiones retinianas periféricas (máx. 8 puntos), lesiones retinianas del polo posterior (máx. 4 puntos), lesiones foveales (máx. 2 puntos) y lesiones del nervio óptico (máx. 2 puntos), con un máximo de 24 puntos. Se utiliza para la evaluación objetiva de la gravedad de los ataques y la determinación de la eficacia del tratamiento6).
Además, la inflamación intraocular puede complicarse con cataratas y glaucoma secundario, causando deterioro visual.
Hallazgos cutáneos: Eritema nudoso que aparece con frecuencia en las piernas, tromboflebitis subcutánea, y erupciones foliculitis-like o acneiformes en la cara, cuello y espalda. Las úlceras genitales son aftosas dolorosas y bien delimitadas, que aparecen con frecuencia en el escroto y pene en hombres, y en los labios mayores y menores en mujeres.
En pacientes pediátricos, se han reportado síntomas oculares graves como uveítis posterior, vasculitis retiniana y papilitis1), y el retraso diagnóstico puede alcanzar un promedio de 11.3 ± 8.5 meses1).
Q¿Cómo se diferencia el hipopión en la enfermedad de Behçet?
A
El hipopión en la enfermedad de Behçet es de naturaleza neutrofílica, por lo que es “fluido”, formando un nivel horizontal claro que se desplaza con los cambios de posición. En cambio, el hipopión en la uveítis asociada a HLA-B27 es más viscoso, no forma nivel y aparece elevado3). Esta diferencia en la consistencia puede ayudar en el diagnóstico.
Se desconoce la causa de la enfermedad de Behçet, pero se sospecha la participación de factores externos como microorganismos patógenos (estreptococos) y factores internos como antecedentes genéticos y anomalías inmunitarias. Las anomalías funcionales de los neutrófilos y las anomalías de citocinas como el TNF-α desempeñan un papel central, provocando reacciones inflamatorias episódicas y recurrentes principalmente en la mucosa oral, ojos, piel y genitales.
Factor de Riesgo
Detalles
HLA-B51
Positivo en aproximadamente el 50% de los pacientes (15% en la población general). Marcador genético principal5)
HLA-A26
Reportado como un alelo de riesgo independiente2)
Región/Etnia
Alta frecuencia a lo largo de la Ruta de la Seda (Mediterráneo, Medio Oriente, Asia Oriental)
Sexo y edad
Aparición entre los 20 y 50 años. La afectación ocular grave es más frecuente en hombres jóvenes.
Los casos pediátricos representan aproximadamente el 1.6–7.7% de todos los casos de enfermedad de Behçet 2), y se ha informado de un empeoramiento de las lesiones oculares en casos HLA-B51 positivos 1).
:::tip Precauciones en la vida diaria
La colchicina debe tomarse a largo plazo para prevenir los ataques oculares. Incluso si los ataques parecen remitir, no suspenda la medicación por su cuenta. Además, se sabe que la fotocoagulación puede desencadenar ataques graves, por lo que es importante discutirlo a fondo con su médico.
:::
El diagnóstico de la enfermedad de Behçet se basa en las guías clínicas del Comité de Investigación de la Enfermedad de Behçet del Ministerio de Salud, Trabajo y Bienestar (1987, revisión menor 2016). Para el diagnóstico basado en síntomas oculares, consulte la Guía Clínica de Lesiones Oculares de la Enfermedad de Behçet (2012) 3).
El tipo completo se define como la aparición de los cuatro síntomas principales durante el curso; el tipo incompleto se define como la aparición de tres síntomas principales, o dos síntomas principales más dos síntomas menores (o síntomas oculares típicos más otro síntoma principal y dos síntomas menores).
Cinco síntomas menores (revisión menor 2016)8): (1) artritis sin deformidad ni rigidez, (2) epididimitis, (3) lesiones gastrointestinales representadas por úlceras ileocecales, (4) lesiones vasculares, (5) lesiones del sistema nervioso central de moderadas a graves
Tipo
Requisito
Tipo completo
Aparecen los cuatro síntomas principales
Tipo incompleto
Tres síntomas principales, o dos síntomas principales + dos síntomas menores
Tipo especial
Tipo intestinal, tipo vascular, tipo neurológico (cuando cumplen los criterios de tipo completo o incompleto y se acompañan de lesiones especiales)
Los criterios diagnósticos internacionales del Grupo Internacional de Estudio de la Enfermedad de Behçet (criterios ISG, 1990) 7) también se utilizan ampliamente a nivel internacional.
Microscopía con lámpara de hendidura: inflamación de la cámara anterior (flare, recuento celular, hipopión). Cuantificado mediante la puntuación BOS24.
Examen de fondo de ojo: opacidad vítrea, vasculitis retiniana, exudados, hemorragia.
Análisis de sangre: recuento de leucocitos, PCR, VSG (marcadores inflamatorios).
Prueba de patergia cutánea (pathergy test): eritema y formación de pústulas tras la inserción intradérmica de una aguja (diagnóstico auxiliar).
En los casos pediátricos, el diagnóstico suele retrasarse; un informe indicó un promedio de 11,3 ± 8,5 meses desde la aparición de los síntomas oculares hasta el diagnóstico 1).
Uveítis asociada a HLA-B27: el hipopión es viscoso e irregular con el centro ligeramente elevado. Casi no hay inflamación del segmento posterior 3). Endoftalmitis fúngica: forma hipopión y opacidades vítreas pero es progresiva. Iritis diabética: rara vez forma hipopión3).
Colchicina oral (uso fuera de indicación): 0.5–1.5 mg/día, generalmente 1 mg/día en dos dosis divididas. Se observa mejoría parcial en aproximadamente el 60% de los pacientes 3). Efectos secundarios: síntomas gastrointestinales como diarrea, teratogenicidad. Se requiere medicación a largo plazo incluso después de que los ataques oculares cedan, y son necesarios análisis de sangre regulares (monitoreo de disfunción hepatorrenal, granulocitopenia, rabdomiólisis).
Ciclosporina (Neoral® 50 mg): Aproximadamente 5 mg/kg/día en dos dosis divididas. Monitorear niveles valle con objetivo de 50–200 ng/mL.
:::caution Efectos secundarios importantes de la ciclosporina
La disfunción renal ocurre con alta frecuencia. Además, se sabe que la enfermedad de Behçet neurológica se desarrolla en aproximadamente el 20% de los casos, por lo que se debe prestar atención a los síntomas neurológicos durante la administración a largo plazo 3). La combinación con colchicina conlleva riesgo de miopatía.
:::
Paso 4: Inhibidores de TNF (casos refractarios/graves)
Aprobación del seguro: Aprobado en 2007 para uveítis retiniana refractaria debida a enfermedad de Behçet
Indicaciones: Uveítis retiniana refractaria resistente a tratamientos existentes o cuando el uso de inmunosupresores es difícil debido a efectos secundarios sistémicos
Posología: 5 mg/kg en infusión intravenosa durante al menos 2 horas. Administrar a las 2 y 6 semanas después de la primera dosis, luego cada 8 semanas. Administrar a través de un filtro de membrana de 1.2 μm o menor. Después de la semana 6, si no hay reacción a la infusión, se puede acortar el tiempo de infusión (pero la velocidad media de infusión no debe exceder 5 mg/kg por hora)
Falta de respuesta primaria y secundaria: Puede ocurrir falta de respuesta primaria (sin efecto desde el inicio) y falta de respuesta secundaria (disminución del efecto durante el tratamiento)
Evaluación pretratamiento (pautas para inhibidores de TNF) 9):
Radiografía de tórax, prueba cutánea de tuberculina; si es fuertemente positiva, QFT/T-SPOT
Lesiones tuberculosas antiguas → Isoniazida oral desde 3 semanas antes hasta 9 meses después de iniciar infliximab
Antígeno HBs, anticuerpo HBs, anticuerpo HBc (monitoreo de reactivación de VHB)
Antígeno HBs positivo → Debe consultar a un hepatólogo. Incluso si es negativo, si el anticuerpo HBs/anticuerpo HBc es positivo (infección pasada) → medición regular de ADN del VHB
Verificar presencia de hepatitis C, HTLV-1, infección por VIH
Enfermedades de base: insuficiencia cardíaca congestiva, enfermedad desmielinizante, neoplasia maligna
Contraindicaciones 9): Infecciones activas incluyendo tuberculosis (infección por micobacterias atípicas, infección por virus de hepatitis B), insuficiencia cardíaca congestiva (NYHA clase III o superior), neoplasia maligna, enfermedades desmielinizantes (ej., esclerosis múltiple)
Monitoreo de efectos secundarios 9): Análisis de sangre periférica regulares (leucocitos, linfocitos) y pruebas bioquímicas (incluyendo PCR). Vigilar la aparición de tuberculosis y neumonía por Pneumocystis (radiografía de tórax, TC, β-D-glucano). Vigilar la reactivación de infección pasada por virus de hepatitis B (ADN-VHB). Vigilar reacciones a la infusión (durante la infusión y 2 horas después). También vigilar hipersensibilidad tardía (dolor muscular, erupción cutánea, fiebre, artralgia que ocurren 3 o más días después de la infusión).
Requisitos del médico y del centro 9): Debe ser un oftalmólogo certificado por la Sociedad Japonesa de Oftalmología y miembro de la Sociedad Japonesa de Inflamación Ocular, y haber completado el e-learning de dicha sociedad. El centro que inicia el tratamiento debe estar registrado en la Sociedad Japonesa de Inflamación Ocular. Se requiere colaboración con internistas experimentados en el manejo de efectos secundarios graves, enfermedades respiratorias/infecciosas e inhibidores de TNF.
Caso representativo: Varón de 32 años, tipo completo HLA-B51 positivo. Incontrolable con ciclosporina, prednisolona y colchicina → introducción de infliximab. En el año anterior al inicio, 3 ataques oculares en ambos ojos combinados → desaparición de ataques en el año posterior al inicio. La agudeza visual corregida mejoró de derecha 1.2/izquierda 0.7 a derecha 1.2/izquierda 0.9. Se mantuvo buena visión incluso después de 3 años y 6 meses3).
Posología: Inyección subcutánea inicial de 80 mg → 40 mg después de 1 semana → luego 40 mg por vía subcutánea cada 2 semanas
Autoinyección: Posible después de suficiente educación y entrenamiento. Incluso después de la autoinyección, es obligatorio el monitoreo de hallazgos oculares y sistémicos cada 2-3 meses
Evaluación previa a la administración y contraindicaciones: Igual que para infliximab
Eficacia para la enfermedad de Behçet: La acumulación de casos está en curso3)
Precauciones sobre la administración sistémica de esteroides
En algunos casos, se desencadenan ataques inflamatorios oculares severos durante la reducción o suspensión, empeorando el pronóstico visual. Generalmente, no se recomiendan los esteroides orales a largo plazo, pero pueden usarse por un período muy corto de aproximadamente una semana cuando los cambios exudativos en la mácula son prominentes3).
Azatioprina: Usada a menudo en el extranjero. A veces considerada de primera línea12)
Inyección intravítrea de triamcinolona acetonida: Proporciona supresión de ataques mientras el fármaco permanece en la cavidad vítrea. Se requieren inyecciones repetidas, y se debe prestar atención a efectos secundarios como cataratas y aumento de la presión intraocular
Interferón alfa-2a: Usado principalmente en Europa. Se ha reportado alta eficacia12)
Precauciones para pacientes quirúrgicos (pautas de uso de inhibidores de TNF)9)
Para cirugía intraocular mínimamente invasiva, la interrupción de los inhibidores de TNF no es una indicación absoluta. Para cirugía extraocular o cirugía altamente invasiva de otros órganos, considere la interrupción (vida media de infliximab aproximadamente 8-9.5 días, adalimumab aproximadamente 14 días).
Glaucoma secundario: Agregar gotas antiglaucomatosas, medicación oral o intravenosa.
Catarata complicada: Se recomienda un período libre de ataques de al menos 6 meses. Cirugía (con posible implante de lente intraocular) después del control de la inflamación.
Edema macular quístico: Inyección subtenoniana de esteroides y/o intensificación de la terapia inmunosupresora.
Fotocoagulación retiniana: Se realiza para vasculitis oclusiva, pero no se realiza de forma casual ya que puede desencadenar ataques inflamatorios oculares graves3).
Q¿En qué pacientes se usa infliximab?
A
Se utiliza en casos refractarios y graves donde los ataques oculares no se controlan con colchicina o ciclosporina. Solo pueden prescribirlo oftalmólogos certificados por la Sociedad Japonesa de Oftalmología y miembros de la Sociedad Japonesa de Inflamación Ocular, que hayan completado el e-learning9). Después de las dosis iniciales en las semanas 0, 2 y 6, se administran dosis de mantenimiento cada 8 semanas. Es obligatorio realizar pruebas de detección de tuberculosis y hepatitis B antes de la administración.
Se cree que la patogenia de la enfermedad de Behçet implica una combinación de predisposición inmunogenética y factores ambientales.
Factores genéticos: HLA-B51 es el marcador genético más fuertemente asociado con la enfermedad de Behçet, positivo en aproximadamente el 50% de los pacientes (frente al 15% en la población general)5). HLA-B15, B27, B40, B44, B52, B57 y A26 también se han identificado como alelos de riesgo independientes2).
Mecanismo inflamatorio: Las anomalías funcionales de los neutrófilos desempeñan un papel central, y la sobreproducción de citocinas como TNF-α desencadena la respuesta inflamatoria. En el ojo, se produce vasculitis oclusiva, que se manifiesta como un aumento de la permeabilidad de los capilares retinianos (fuga de fluoresceína en forma de helecho en la angiografía fluoresceínica). Durante las exacerbaciones agudas, la infiltración de leucocitos (neutrófilos) y la isquemia conducen a la formación de exudados blancos.
Características de la inflamación no granulomatosa: Iridociclitis no granulomatosa, donde las células inflamatorias no forman grumos → precipitados queráticos en grasa de carnero. Esta es una característica importante para la diferenciación de uveítis granulomatosa como la sarcoidosis y la enfermedad de Harada3).
Ataques inflamatorios recurrentes: Los ataques oculares repetidos conducen a atrofia retiniana y atrofia del nervio óptico, lo que resulta en un deterioro visual grave. El hecho de que el hipopión sea neutrofílico y tenga una consistencia fina y acuosa también refleja el mecanismo inflamatorio dominado por neutrófilos.
7. Investigación más reciente y perspectivas futuras
En los casos representativos de las guías, se ha informado de un curso favorable durante 3 años y 6 meses 3). La introducción de infliximab reduce notablemente la frecuencia de los ataques oculares y mantiene o mejora la agudeza visual10). Una revisión sobre la administración a largo plazo de infliximab para la enfermedad de Behçet refractaria en Japón también muestra una alta eficacia en la supresión de ataques y el mantenimiento de la visión 11).
La comparación de pacientes de las décadas de 1980 y 1990 (Yoshida 2004) confirma una tendencia hacia una enfermedad más leve 4). La prevalencia disminuyó del 6,2% al 3,9% entre 2002 y 2009 5). En paralelo con la difusión de los agentes biológicos, la incidencia de deterioro visual grave también ha disminuido.
Los inhibidores del TNF (infliximab, adalimumab) han demostrado eficacia en múltiples enfermedades, incluida la uveítis no infecciosa asociada a la enfermedad de Behçet 12). Incluso en casos pediátricos, se ha informado de un buen mantenimiento de la agudeza visual con tratamiento agresivo que incluye infliximab1). La evidencia de adalimumab para la enfermedad de Behçet se está acumulando, y se esperan resultados de futuras investigaciones 9).
:::danger Aviso legal
Este artículo tiene como objetivo proporcionar información médica y no indica diagnóstico o tratamiento individual. Las decisiones sobre el tratamiento deben seguir siempre las instrucciones de un especialista. La dosis y la administración de los medicamentos varían según la condición individual, por lo que consulte a su médico para la prescripción real.
:::
Casem Azri, Perrine Dusser, Laura Eid, Emmanuel Barreau, Isabelle Kone-Paut, Charlotte Borocco, Caroline Galeotti, Sami Saad, et al. Ocular involvement in pediatric Behçet’s disease: is it different than in adults? (a short case series and mini review). BMC Ophthalmol. 2023;23(1). doi:10.1186/s12886-023-03197-5.
Xin Yao, Xing-Ning Wang, Jian-Ming Lai. Pediatric Behçet’s disease with cardiac valvular lesions: A case-based review. Science Progress. 2023;106(2). doi:10.1177/00368504231173404.
Yoshida A, Kawashima H, Motoyama Y, Shibui H, Kaburaki T, Shimizu K, Ando K, Hijikata K, Izawa Y, Hayashi K, Numaga J, Fujino Y, Masuda K, Araie M.. Comparison of patients with Behçet’s disease in the 1980s and 1990s. Ophthalmology. 2004;111(4):810-815. doi:10.1016/j.ophtha.2003.07.018. PMID:15051217.
Kaburaki T, Namba K, Sonoda KH, Kezuka T, Keino H, Fukuhara T, Kamoi K, Nakai K, Mizuki N, Ohguro N, Ocular Behçet Disease Research Group of Japan.. Behçet’s disease ocular attack score 24: evaluation of ocular disease activity before and after initiation of infliximab. Jpn J Ophthalmol. 2014;58(2):120-130. doi:10.1007/s10384-013-0294-0. PMID:24482146.
INTERNATIONALSTUDYGROUPFORBEHC. Criteria for diagnosis of Behcet’s disease. The Lancet. 1990;335(8697). doi:10.1016/0140-6736(90)92643-v.
Ohno S, Nakamura S, Hori S, Shimakawa M, Kawashima H, Mochizuki M, Sugita S, Ueno S, Yoshizaki K, Inaba G.. Efficacy, safety, and pharmacokinetics of multiple administration of infliximab in Behçet’s disease with refractory uveoretinitis. J Rheumatol. 2004;31(7):1362-1368. PMID:15229958.
Namba K, Goto H, Kaburaki T, et al. A major review: current aspects of ocular Behçet’s disease in Japan. Ocul Immunol Inflamm. 2015;23(Suppl 1):S1-S23. doi:10.3109/09273948.2014.981547.
Pasadhika S, Rosenbaum JT.. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics. 2014;8:67-81. doi:10.2147/btt.s41477. PMID:24600203; PMCID:PMC3933243.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.