Behçet’s disease is a refractory systemic inflammatory disease of unknown cause characterized by four major symptoms: oral aphthous ulcers, ocular lesions, genital ulcers, and skin lesions. Inflammation is acute and transient, but the disease is characterized by repeated relapses. It occurs in young to middle-aged adults (20s–50s) with a male-to-female ratio of approximately 1:1. Uveitis occurs in about 70% of male patients and 45% of female patients, with severe cases more common in young men.
It is frequently observed along the Silk Road from the Mediterranean to China and Japan. About 50% of patients are positive for HLA-B51 (15% in the general population)5), and HLA-A26 (A*2601) has also been reported as an independent risk allele. Immunogenetic predisposition and environmental factors are thought to be involved in the onset. This disease once had a high blindness rate, but advances in immunosuppressants and biologics have reduced the blindness rate.
Behçet’s disease is registered as a designated intractable disease (No. 56)8).
Disease characteristics
Age of onset: Young to middle-aged adults (20s–50s)
Sex difference: Male-to-female ratio approximately 1:1. Ocular lesions are often severe in young men.
Geographic distribution: High frequency along the Silk Road (Mediterranean to Middle East to East Asia).
Genetic background: HLA-B51 positive (about 50% of patients), HLA-A26 is also an independent risk factor 5).
4 Major Symptoms
Oral aphthous ulcers: Painful recurrent ulcers. Often the initial symptom.
The proportion of Behçet’s disease among all uveitis cases has been decreasing over time. In the 2002 epidemiological survey, it was 6.2% (3rd most common), but in 2009 it decreased to 3.9% (6th most common) 5). In addition to a decrease in the number of patients, a milder disease course has also been reported 4).
QWhat kind of disease is Behçet's disease?
A
Behçet’s disease is a systemic inflammatory disease that recurs repeatedly, characterized by four major symptoms: oral aphthous ulcers, ocular symptoms, genital ulcers, and skin symptoms. The cause is unknown, but an autoimmune mechanism is suspected. It is more common in regions along the Silk Road, and genetic background (HLA-B51) and environmental factors are thought to be involved. Ocular lesions are particularly important and can lead to blindness without appropriate treatment.
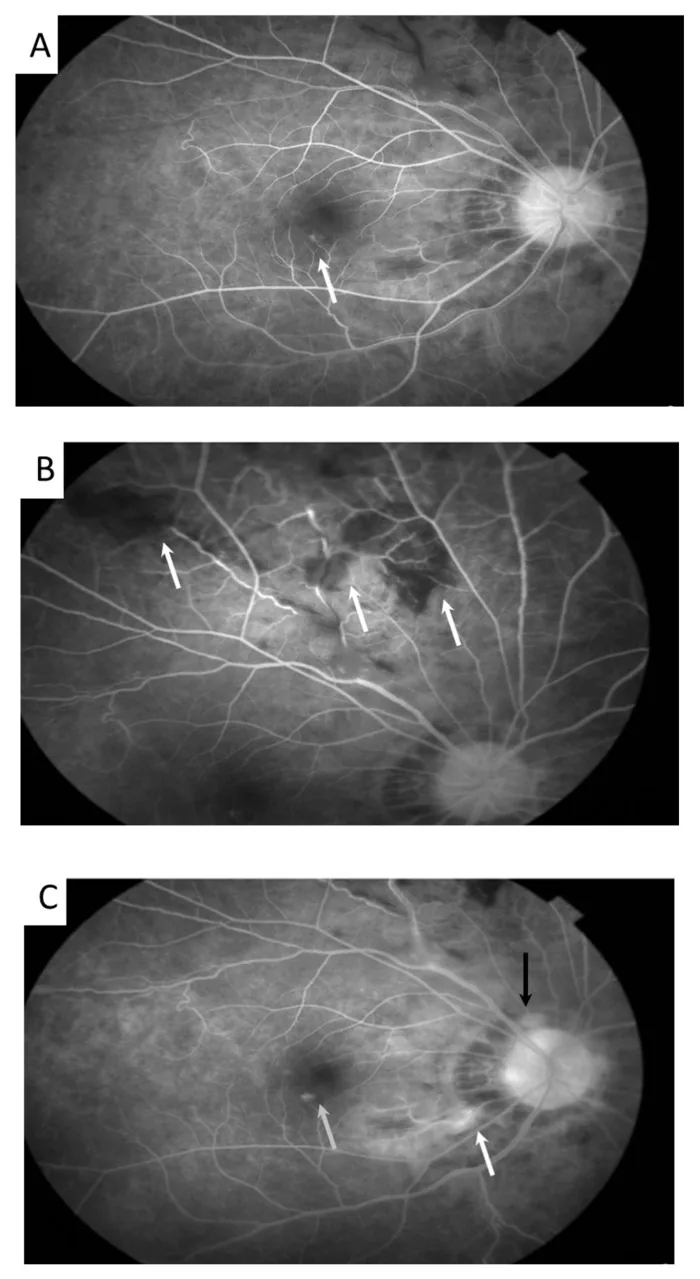
Meng PP, et al. Use of Ultra-Widefield Fluorescein Angiography to Guide the Treatment to Idiopathic Retinal Vasculitis, Aneurysms, and Neuroretinitis-Case Report and Literature Review. Medicina (Kaunas). 2022. Figure 2. PMCID: PMC9611749. License: CC BY.
In autofluorescence imaging, aneurysmal dilation of retinal arterioles (arrow) and venous tortuosity are observed. This corresponds to the findings of retinal vasculitis discussed in the section “2. Main Symptoms and Clinical Findings”.
Ocular subjective symptoms include blurred vision, decreased visual acuity, and floaters during inflammatory attacks. Inflammation often recurs in one eye at different times, eventually becoming bilateral in about 90% of cases. In severe cases, attacks may occur several times a month, while in mild cases, attacks occur about once a year and continue for several years to over a decade. Retinitis localized to the macula may recur, causing irreversible vision loss.
Systemic symptoms mainly include oral aphthous ulcers (about 90%), skin lesions (about 75%), and genital ulcers (about 50%). Oral aphthous ulcers commonly occur on the tongue, buccal mucosa, lips, and gums, and are painful ulcers with surrounding redness. They are almost always present in this disease and are often the initial symptom. They heal without scarring within 10 days but recur repeatedly.
Secondary symptoms include arthritis without deformity or stiffness, epididymitis, gastrointestinal Behçet’s disease (ileocecal ulcers), vascular Behçet’s disease (vasculitis), and neuro-Behçet’s disease (meningoencephalitis). Since extraocular symptoms and ocular findings are not always linked, it is important to evaluate systemic status in addition to ophthalmologic observation3).
The following two ocular symptoms recur paroxysmally.
(1) Acute iridocyclitis, sometimes with hypopyon.
(2) Diffuse vitreous opacities, retinochoroiditis, and retinal vasculitis.
Early and active phase findings of ocular attacks: Paroxysmal ocular inflammation. Fine keratic precipitates, anterior chamber inflammatory cells, and hypopyon. Fibrin exudation in the anterior chamber is usually absent. No iris or angle nodules (non-granulomatous). In the posterior segment, vitreous opacities, retinal exudates, and hemorrhages characteristically resolve relatively quickly (1–2 weeks)3).
Late findings3): Retinochoroidal atrophy, retinal vascular sheathing, optic disc pallor → leading to severe visual dysfunction.
Hypopyon in Behçet’s disease is fluid due to predominant neutrophil infiltration, forming a clean horizontal level. It characteristically shifts easily with changes in body position, differing from the viscous hypopyon of HLA-B27-associated uveitis3). Keratic precipitates are fine and dust-like, showing non-granulomatous findings.
During acute exacerbation of retinal uveitis, vitreous opacities, retinal vasculitis, white retinal exudates, and hemorrhages are observed. White exudates are due to leukocyte infiltration and ischemic swelling of nerve fibers, and characteristically resolve relatively quickly (within about a week). Occlusive vasculitis may cause hemorrhages resembling branch retinal vein occlusion.
Fluorescein angiography often reveals extensive, vigorous leakage from retinal capillaries (fern-like leakage) even when no ocular attack is present, which is considered characteristic of this disease.
A scoring system that quantifies the degree of inflammation for each ocular attack. It consists of six items: anterior chamber inflammation (max 4 points), vitreous opacity (max 4 points), peripheral retinal lesions (max 8 points), posterior pole retinal lesions (max 4 points), foveal lesions (max 2 points), and optic nerve lesions (max 2 points), with a maximum total of 24 points. It is used for objective assessment of attack severity and evaluation of treatment efficacy6).
Additionally, intraocular inflammation may be accompanied by complicated cataract and secondary glaucoma, which can cause visual impairment.
Skin findings: Erythema nodosum commonly occurring on the lower legs, subcutaneous thrombophlebitis, and folliculitis-like or acneiform rashes on the face, neck, and back. Genital ulcers are painful, well-demarcated aphthous ulcers, commonly occurring on the scrotum and penis in men, and on the labia majora and minora in women.
In pediatric patients, severe ocular symptoms such as posterior uveitis, retinal vasculitis, and papillitis have been reported1), and diagnostic delay can average 11.3 ± 8.5 months1).
QHow can hypopyon in Behçet's disease be differentiated?
A
Hypopyon in Behçet’s disease is neutrophilic, thus “watery,” forming a clear horizontal level that shifts with changes in body position. In contrast, hypopyon in HLA-B27-associated uveitis is more viscous, does not form a level, and appears raised3). This difference in consistency can aid in diagnosis.
The cause of Behçet’s disease is unknown, but external factors such as involvement of pathogenic microorganisms like streptococci, and internal factors such as genetic background and immune abnormalities are suspected. Functional abnormalities of neutrophils and abnormalities of cytokines including TNF-α play a central role, leading to episodic and recurrent inflammatory reactions mainly in the oral mucosa, eyes, skin, and genitalia.
Risk Factor
Details
HLA-B51
Positive in approximately 50% of patients (15% in the general population). A major genetic marker5)
HLA-A26
Reported as an independent risk allele2)
Region/Ethnicity
High frequency along the Silk Road (Mediterranean to Middle East to East Asia)
Sex and age
Onset between 20s and 50s. Severe ocular involvement is more common in young men.
Pediatric cases account for approximately 1.6–7.7% of all Behçet’s disease cases 2), and severe ocular involvement has been reported in HLA-B51-positive cases 1).
:::tip Daily life precautions
Colchicine must be taken long-term to prevent ocular attacks. Even if attacks seem to subside, do not stop medication on your own. Also, photocoagulation therapy is known to sometimes trigger severe attacks, so it is important to discuss thoroughly with your doctor.
:::
The diagnosis of Behçet’s disease is based on the clinical guidelines of the Ministry of Health, Labour and Welfare’s Behçet’s Disease Research Committee (1987, minor revision 2016). For diagnosis based on ocular symptoms, refer to the Behçet’s Disease Ocular Lesion Clinical Guidelines (2012) 3).
Complete type is defined as the appearance of all four major symptoms during the course; incomplete type is defined as the appearance of three major symptoms, or two major symptoms plus two minor symptoms (or typical ocular symptoms plus one other major symptom and two minor symptoms).
Five minor symptoms (2016 minor revision)8): (1) arthritis without deformity or stiffness, (2) epididymitis, (3) gastrointestinal lesions represented by ileocecal ulcers, (4) vascular lesions, (5) moderate to severe central nervous system lesions
Type
Requirement
Complete type
All four major symptoms appear
Incomplete type
Three major symptoms, or two major symptoms + two minor symptoms
Special type
Intestinal type, vascular type, neurological type (when meeting criteria for complete or incomplete type and accompanied by special lesions)
The International Diagnostic Criteria of the International Study Group for Behçet’s Disease (ISG criteria, 1990) 7) are also widely used internationally.
HLA-B27-associated uveitis: hypopyon is viscous and irregular with a slightly raised center. There is almost no posterior segment inflammation 3). Fungal endophthalmitis: forms hypopyon and vitreous opacities but is progressive. Diabetic iritis: rarely forms a hypopyon3).
Oral colchicine (off-label use): 0.5–1.5 mg/day, usually 1 mg/day in two divided doses. Partial improvement is seen in about 60% of patients 3). Side effects: gastrointestinal symptoms such as diarrhea, teratogenicity. Long-term medication is required even after ocular attacks subside, and regular blood tests (monitoring for hepatorenal dysfunction, granulocytopenia, rhabdomyolysis) are necessary.
Cyclosporine (Neoral® 50 mg): Approximately 5 mg/kg/day in two divided doses. Monitor trough levels targeting 50–200 ng/mL.
:::caution Important side effects of cyclosporine
Renal dysfunction occurs at a high rate. Furthermore, it is known that neuro-Behçet’s disease develops in about 20% of cases, so attention to neurological symptoms is necessary during long-term administration 3). Combination with colchicine carries a risk of myopathy.
:::
Insurance approval: Approved in 2007 for refractory retinal uveitis due to Behçet’s disease
Indications: Refractory retinal uveitis resistant to existing treatments or where immunosuppressants are difficult to use due to systemic side effects
Dosage: 5 mg/kg intravenous infusion over at least 2 hours. Administered at weeks 2 and 6 after the first dose, then every 8 weeks. Administer through a membrane filter of 1.2 μm or smaller. After week 6, if no infusion reaction occurs, infusion time may be shortened (but the average infusion rate should not exceed 5 mg/kg per hour)
Primary and secondary non-response: Primary non-response (no effect from the start) and secondary non-response (diminished effect during treatment) may occur
HBs antigen positive → Must consult a hepatologist. Even if negative, if HBs antibody/HBc antibody positive (past infection) → regular HBV DNA measurement
Contraindications 9): Active infections including tuberculosis (atypical mycobacterial infection, hepatitis B virus infection), congestive heart failure (NYHA class III or higher), malignancy, demyelinating diseases (e.g., multiple sclerosis)
Side effect monitoring 9): Regular peripheral blood tests (white blood cells, lymphocytes) and biochemical tests (including CRP). Monitor for tuberculosis and Pneumocystis pneumonia (chest X-ray, CT, β-D-glucan). Monitor for reactivation of past hepatitis B virus infection (HBV-DNA). Monitor for infusion reactions (during infusion and for 2 hours after). Also monitor for delayed hypersensitivity (muscle pain, rash, fever, joint pain occurring 3 or more days after infusion).
Physician and facility requirements 9): Must be a board-certified ophthalmologist of the Japanese Ophthalmological Society and a member of the Japanese Society of Ocular Inflammation, and have completed the society’s e-learning. The initiating facility must be registered with the Japanese Society of Ocular Inflammation. Collaboration with internists experienced in managing severe side effects, respiratory/infectious diseases, and TNF inhibitors is required.
Representative case: 32-year-old male, HLA-B51-positive complete type. Uncontrollable with cyclosporine, prednisolone, and colchicine → infliximab introduced. In the year before initiation, 3 ocular attacks in both eyes combined → attacks disappeared in the year after initiation. Corrected visual acuity improved from right 1.2/left 0.7 to right 1.2/left 0.9. Good vision maintained even after 3 years and 6 months3).
Insurance approval: Approved in 2016 for non-infectious intermediate, posterior, and panuveitis
Dosage: Initial 80mg subcutaneous injection → 40mg after 1 week → then 40mg subcutaneously every 2 weeks
Self-injection: Possible after sufficient education and training. Even after self-injection, monitoring of ocular and systemic findings every 2-3 months is mandatory
Pre-administration screening and contraindications: Same as for infliximab
Efficacy for Behçet’s disease: Case accumulation is ongoing3)
In some cases, severe ocular inflammatory attacks are triggered during dose reduction or discontinuation, worsening visual prognosis. Generally, long-term oral steroids are not recommended, but they may be used for a very short period of about one week when exudative changes in the macula are prominent3).
Azathioprine: Often used overseas. Sometimes considered first-line12)
Intravitreal triamcinolone acetonide injection: Provides attack suppression while the drug remains in the vitreous cavity. Repeated injections are necessary, and attention should be paid to side effects such as cataracts and increased intraocular pressure
Interferon-alpha-2a: Used mainly in Europe. High efficacy has been reported12)
Precautions for surgical patients (guidelines for TNF inhibitor use)9)
For minimally invasive intraocular surgery, discontinuation of TNF inhibitors is not an absolute indication. For extraocular surgery or highly invasive surgery of other organs, consider discontinuation (infliximab half-life approximately 8-9.5 days, adalimumab approximately 14 days).
Complicated cataract: A seizure-free period of at least 6 months is desirable. Surgery (with intraocular lens implantation possible) after inflammation control.
Retinal photocoagulation: Performed for occlusive vasculitis, but not performed casually as it may trigger severe ocular inflammatory attacks3).
QIn which patients is infliximab used?
A
It is used in refractory and severe cases where ocular attacks cannot be controlled with colchicine or cyclosporine. Only ophthalmologists who are certified by the Japanese Ophthalmological Society and members of the Japanese Society for Ocular Inflammation, and who have completed e-learning, can prescribe it9). After initial doses at weeks 0, 2, and 6, maintenance doses are given every 8 weeks. Screening for tuberculosis and hepatitis B is mandatory before administration.
The pathogenesis of Behçet’s disease is thought to involve a combination of immunogenetic predisposition and environmental factors.
Genetic factors: HLA-B51 is the strongest genetic marker associated with Behçet’s disease, positive in about 50% of patients (vs. 15% in the general population)5). HLA-B15, B27, B40, B44, B52, B57, and A26 have also been identified as independent risk alleles2).
Inflammatory mechanism: Functional abnormalities of neutrophils play a central role, and overproduction of cytokines such as TNF-α triggers inflammatory responses. In the eye, occlusive vasculitis occurs, manifesting as increased permeability of retinal capillaries (fern-like fluorescence leakage on fluorescein angiography). During acute exacerbations, infiltration of leukocytes (neutrophils) and ischemia lead to the formation of white exudates.
Characteristics of non-granulomatous inflammation: Non-granulomatous iridocyclitis, where inflammatory cells do not form clumps → mutton-fat keratic precipitates. This is an important feature for differentiation from granulomatous uveitis such as sarcoidosis and Vogt-Koyanagi-Harada disease3).
Recurrent inflammatory attacks: Repeated ocular attacks lead to retinal atrophy and optic nerve atrophy, resulting in severe visual impairment. The fact that hypopyon is neutrophilic and has a thin, watery consistency also reflects the neutrophil-dominated inflammatory mechanism.
In representative cases from the guidelines, a favorable course over 3 years and 6 months has been reported 3). The introduction of infliximab significantly reduces the frequency of ocular attacks and maintains or improves visual acuity10). A review of long-term infliximab administration for refractory Behçet’s disease in Japan also demonstrates high efficacy in suppressing attacks and maintaining vision 11).
Comparison of patients from the 1980s and 1990s (Yoshida 2004) confirms a trend toward milder disease 4). The prevalence decreased from 6.2% to 3.9% between 2002 and 2009 5). Along with the widespread use of biologics, the incidence of severe visual impairment has also declined.
TNF inhibitors (infliximab, adalimumab) have been reported effective in multiple diseases including non-infectious uveitis associated with Behçet’s disease 12). Even in pediatric cases, aggressive treatment including infliximab has been reported to maintain good visual acuity1). Evidence for adalimumab in Behçet’s disease is accumulating, and future research results are anticipated 9).
:::danger Disclaimer
This article is intended to provide medical information and does not indicate individual diagnosis or treatment. Decisions regarding treatment should always follow the instructions of a specialist. Dosage and administration of medications vary depending on individual conditions, so please consult your physician for actual prescriptions.
:::
Casem Azri, Perrine Dusser, Laura Eid, Emmanuel Barreau, Isabelle Kone-Paut, Charlotte Borocco, Caroline Galeotti, Sami Saad, et al. Ocular involvement in pediatric Behçet’s disease: is it different than in adults? (a short case series and mini review). BMC Ophthalmol. 2023;23(1). doi:10.1186/s12886-023-03197-5.
Xin Yao, Xing-Ning Wang, Jian-Ming Lai. Pediatric Behçet’s disease with cardiac valvular lesions: A case-based review. Science Progress. 2023;106(2). doi:10.1177/00368504231173404.
Yoshida A, Kawashima H, Motoyama Y, Shibui H, Kaburaki T, Shimizu K, Ando K, Hijikata K, Izawa Y, Hayashi K, Numaga J, Fujino Y, Masuda K, Araie M.. Comparison of patients with Behçet’s disease in the 1980s and 1990s. Ophthalmology. 2004;111(4):810-815. doi:10.1016/j.ophtha.2003.07.018. PMID:15051217.
Kaburaki T, Namba K, Sonoda KH, Kezuka T, Keino H, Fukuhara T, Kamoi K, Nakai K, Mizuki N, Ohguro N, Ocular Behçet Disease Research Group of Japan.. Behçet’s disease ocular attack score 24: evaluation of ocular disease activity before and after initiation of infliximab. Jpn J Ophthalmol. 2014;58(2):120-130. doi:10.1007/s10384-013-0294-0. PMID:24482146.
INTERNATIONALSTUDYGROUPFORBEHC. Criteria for diagnosis of Behcet’s disease. The Lancet. 1990;335(8697). doi:10.1016/0140-6736(90)92643-v.
Ohno S, Nakamura S, Hori S, Shimakawa M, Kawashima H, Mochizuki M, Sugita S, Ueno S, Yoshizaki K, Inaba G.. Efficacy, safety, and pharmacokinetics of multiple administration of infliximab in Behçet’s disease with refractory uveoretinitis. J Rheumatol. 2004;31(7):1362-1368. PMID:15229958.
Namba K, Goto H, Kaburaki T, et al. A major review: current aspects of ocular Behçet’s disease in Japan. Ocul Immunol Inflamm. 2015;23(Suppl 1):S1-S23. doi:10.3109/09273948.2014.981547.
Pasadhika S, Rosenbaum JT.. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics. 2014;8:67-81. doi:10.2147/btt.s41477. PMID:24600203; PMCID:PMC3933243.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.