Prodromal Stage
Timing: 1–2 weeks before ocular symptoms appear
Symptoms: Scalp tingling, headache (64.7%), tinnitus (24.7%), malaise (21.1%), flu-like symptoms2)

Vogt-Koyanagi-Harada disease (VKH disease) is a T-cell-mediated autoimmune disease against melanin proteins. In the onset phase, it presents with uveitis, meningeal irritation symptoms such as headache, and hearing loss. As inflammation persists, skin manifestations such as vitiligo, alopecia, and poliosis appear. Inflammatory lesions occur throughout the body where melanocytes are distributed: eyes, hair, skin, inner ear, and meninges.
Approximately half of patients have flu-like symptoms within 2 weeks before onset, suggesting that viral infection may trigger the disease through immunological cross-reactivity (molecular mimicry). Involvement of Epstein-Barr virus and cytomegalovirus has been suggested. Visual prognosis is generally good, but some cases progress to severe visual impairment due to persistent inflammation, making early diagnosis and initial treatment important.
VKH is the second most common cause of uveitis. In a 2002 epidemiological survey, 205 cases (6.7%) of VKH were reported, and in a 2009 survey, 267 cases (7.0%) were reported, both ranking second after sarcoidosis1). The typical age of onset is between 20 and 50 years, with a slight female predominance. HLA-DR4 positivity is found in 80% of patients (compared to 25% in the normal Japanese population), and it is more common in people of color (Asians, Hispanics, Native Americans) and rare in Caucasians. Among 93 VKH patients enrolled in the FAST trial, 71% were female, with a median age of 35–38 years2). It is most common in East Asia but also occurs in the Pacific regions of North and South America3).
It is more common in people of color (Asians, Hispanics, Native Americans) and rare in Caucasians. It tends to occur in HLA-DR4-positive individuals and those aged 20–50 years, with a slight female predominance. It is thought to develop from a combination of genetic predisposition (HLA-DRB1*0405) and some environmental trigger3).
Prodromal Stage
Timing: 1–2 weeks before ocular symptoms appear
Symptoms: Scalp tingling, headache (64.7%), tinnitus (24.7%), malaise (21.1%), flu-like symptoms2)
Acute Stage (Ocular Onset)
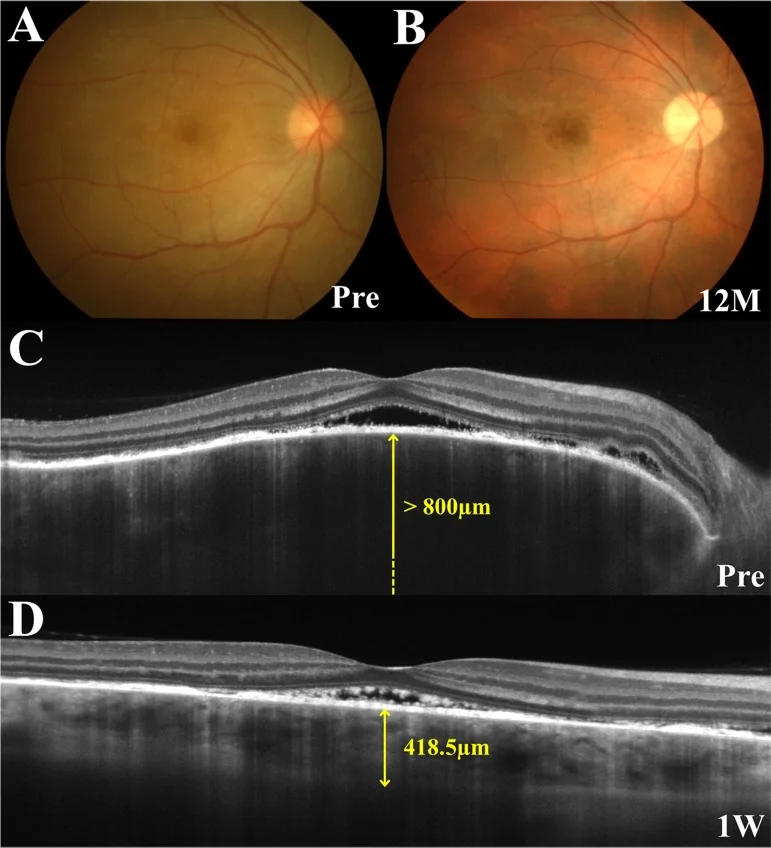
Acute vision loss: Bilateral serous retinal detachment (57.6% of acute VKH)2)
Mild anterior chamber inflammation and vitreous haze, optic disc hyperemia and swelling (about 70%)
Choroidal folds: In severe cases, choroidal detachment and shallow anterior chamber
Chronic Stage (Recurrent Stage)
Granulomatous inflammation of the anterior segment is predominant
Mutton-fat KP, Koeppe/Busacca nodules, posterior synechiae
Sunset glow fundus appears in 33.3% of chronic VKH2)
Late (convalescent) stage
Sunset glow fundus: loss of choroidal melanin pigment
Sugiura sign: depigmentation of the limbus (about 1 month after onset)
Skin manifestations (about 20%): vitiligo, alopecia, poliosis (after several months)
Macular pigment accumulation, scattered chorioretinal depigmented spots
In the prodromal stage, scalp paresthesia, headache, and tinnitus appear, followed 1–2 weeks later by acute bilateral or sequential vision loss and blurred vision. On OCT, septal structures are characteristically observed within the retinal detachment in the acute phase.
Meningeal irritation (headache, nuchal rigidity) occurs in most cases early in the disease. Sensorineural hearing loss (often subclinical but detected on testing). Skin manifestations (vitiligo, alopecia, poliosis) appear several months after onset and are seen in about 20% of patients.
During recurrence, serous retinal detachment of the posterior segment, as seen in the initial episode, is less common, and granulomatous inflammation of the anterior segment predominates. Mutton-fat keratic precipitates (KP), Koeppe nodules, and Busacca nodules are observed, often leading to posterior synechiae. Prodromal symptoms may include ocular redness and blurred vision.
The core of the pathology is a CD4-positive T-cell autoimmune response against melanin proteins (tyrosinase family: tyrosinase, TRP-1, TRP-2, gp100) 3). The target organs are melanocytes in the uvea (choroid), central nervous system (meninges), inner ear, and skin, with granulomatous inflammation of the choroid being the initial main lesion.
A strong association with HLA-DR4 (especially DRB10405) is known as a genetic predisposition 3). Linkage disequilibrium with HLA-DPB10501 has also been reported 3). There is a hypothesis that viral infections (EBV, CMV) trigger autoimmunity through molecular mimicry.
Main risk factors:
| Type | Criteria |
|---|---|
| Complete VKH disease | Ocular findings + neurological/auditory findings + skin findings, all present |
| Incomplete VKH disease | Ocular findings present, lacking either neurological/auditory findings or skin findings |
| Probable VKH disease | Ocular findings only (isolated uveitis) |
In the early stage of onset, skin findings are not observed, so most cases are incomplete type. Later, when skin findings appear, it becomes complete type. Bilateral serous retinal detachment in the early stage is characteristic, and diagnosis is easy if there are prodromal symptoms such as headache or extraocular symptoms such as tinnitus 1). In atypical cases (optic disc edema type, unilateral cases), cerebrospinal fluid examination is decisive for diagnosis.
Cerebrospinal fluid examination: Lymphocyte-predominant pleocytosis. Persists for up to 8 weeks. Most useful for confirming diagnosis.
Fluorescein angiography (FA): Early phase shows patchy hypofluorescence due to delayed choroidal filling → mid-phase shows granular hyperfluorescence (pinpoint leakage) → late phase shows dye pooling in the area of serous retinal detachment. Optic disc hyperfluorescence (approximately 70%) 1).
ICG angiography: Early phase shows patchy delayed choroidal filling and dye leakage from choroidal vessels; mid to late phase shows scattered hypofluorescent spots. Blurring of medium and large vessels due to choroidal circulatory disturbance is also characteristic.
OCT: Detection and monitoring of serous retinal detachment. Marked choroidal thickening in the early stage. EDI-OCT allows detailed observation of the choroidal cross-section. Fibrin-like membranous structures or septa may be observed within the serous retinal detachment 1).
Ultrasound B-mode: Findings of choroidal thickening. Useful for differentiation from posterior scleritis 1).
HLA class II: HLA-DR4 (adjunctive test). Positive rate 80%, but specificity is low (25% positive in normal individuals).
ERG (electroretinogram): In the chronic phase, amplitudes of photopic and scotopic responses decrease 3).
| Differential Disease | Differentiating Points |
|---|---|
| Posterior scleritis | Unilateral, T-sign on ultrasound, pain on eye movement/tenderness |
| Multiple posterior pigment epitheliopathy (MPPE) | Unilateral, shifting bullous serous retinal detachment |
| Acute posterior multifocal placoid pigment epitheliopathy (APMPPE) | FA reversal phenomenon (early hyperfluorescence → late hypofluorescence), young age, preceding infection |
| Central serous chorioretinopathy (CSCR) | Unilateral, male predominance, steroids as exacerbating factor3) |
| Sympathetic ophthalmia | Differentiated by history of penetrating ocular trauma or intraocular surgery. Pathologically considered the same disease1) |
| Idiopathic uveal effusion syndrome | Association with nanophthalmos, caution for angle closure |
It is not mandatory, but it can be decisive in atypical cases that are difficult to diagnose (optic disc edema type, unilateral cases). Since lymphocytic pleocytosis persists for up to 8 weeks, performing it at an appropriate time after onset improves diagnostic accuracy. In typical cases with obvious bilateral serous retinal detachment, diagnosis can often be made based on clinical findings alone.
Steroid pulse therapy followed by oral high-dose tapering is standard. If left untreated, many cases develop recurrence or persistence leading to severe visual impairment. Since current medicine has no indicator to distinguish cases that heal spontaneously from those that recur or persist, high-dose steroid administration is recommended when possible.
Guideline representative case prescription example1):
Oral tapering schedule (Predonine tablets 5 mg):
| Dose | Duration |
|---|---|
| 200 mg/day | 2 days |
| 150 mg/day | 2 days |
| 100 mg/day | 2 days |
| 80 mg/day | 2 days |
| 60 mg/day | 4 days |
| 40 mg/day | 10 days |
| 30 mg/day | 2 weeks |
| 20 mg/day | 4 weeks |
| 15 mg/day | 4 weeks |
| 10 mg/day | 4 weeks |
| 5 mg/day | 4 weeks |
| 5 mg/day (every other day) | 4 weeks |
Tapering should be done slowly, and even without recurrence, discontinuation should take at least 6 months. Treatment lasting less than 6 months has a recurrence rate of 58.8%, while treatment lasting 6 months or more reduces it to 11.1% 5). If recurrence occurs, increase the steroid dose and taper more slowly than before. With steroid monotherapy, 44% relapse and 59% develop sunset glow fundus 2).
Treatment dose for pediatric patients: In children of a certain age and weight, either methylprednisolone 500 mg/day IV for 3 days (pulse therapy) or oral prednisolone starting at 0.5–1.0 mg/kg/day with gradual tapering is used. Taper while monitoring for side effects.
Cyclosporine (Neoral®): 3 mg/kg/day (for 60 kg body weight: 180 mg/day in two divided doses). Regular trough level monitoring is required. Expected to reduce steroid dose. Used in refractory cases 1). Monitor for side effects such as increased susceptibility to infections, renal dysfunction, and hepatic dysfunction.
Methotrexate (MTX): 25 mg/week orally. Start at 15 mg/week for 2 weeks, then increase 2).
Mycophenolate mofetil (MMF): 1.5 g twice daily orally. Start at 500 mg twice daily and titrate up 2).
The FAST trial (NCT01829295) is an RCT for non-infectious uveitis (subanalysis of 93 VKH cases out of 216). 49 patients were randomized to MTX and 44 to MMF, starting with prednisone 1 mg/kg/day (max 60 mg/day) and tapering (target ≤7.5 mg/day at 6 months).
6-month primary outcome (VKH subanalysis):
| Outcome | MTX group | MMF group | P value |
|---|---|---|---|
| Treatment success rate | 80.4% (37/46) | 64.1% (25/39) | 0.10 |
| Reduction in central foveal thickness | −62.5 μm | −4.0 μm | 0.003 |
| SRD resolution rate | 86.3% | 64.1% | 0.02 |
| Visual improvement | Equivalent | Equivalent | 0.78 |
Overall treatment success rate was 74.7% (62/85 cases). In acute VKH, MTX was superior in reducing CST and resolving SRD. At 12 months, 91.3% of the MMF group continued successful treatment, and about half (MTX 50.0%, MMF 56.5%) achieved complete prednisone withdrawal 2). Switching from MMF to MTX after initial treatment failure was successful in 81.8% of cases 2). With initial MMF combination, 93% of acute VKH patients maintained 20/20 vision, and all cases had no recurrence or sunset glow fundus formation 2).
For cases where systemic steroid administration is difficult (elderly, pregnant, diabetes, history of psychiatric disease), consider posterior sub-Tenon injection of triamcinolone acetonide 1).
Complicated cataract: Steroid cataract occurs at a high rate due to heavy steroid use. In completely remitted cases, the risk is similar to normal surgery. IOL implantation is also not problematic. If filtration surgery may be needed later, choose superior conjunctival preservation and corneal incision.
Secondary glaucoma / steroid glaucoma: Use antihypertensive eye drops (beta-blockers, PG agents, carbonic anhydrase inhibitors) → oral CAI → intravenous D-mannitol in order. Perform trabeculotomy (effective for steroid glaucoma), and if insufficient, trabeculectomy.
Even without recurrence, it is recommended to taper slowly over at least 6 months. Discontinuation within less than 6 months results in a high recurrence rate of about 58.8%, while continuation for 6 months or more reduces it to 11.1% 5). In case of recurrence, taper more slowly than the previous time and consider adding immunosuppressive drugs.
Cyclosporine (Neoral® 3 mg/kg/day) is used as a steroid-sparing agent in refractory cases 1). The FAST trial confirmed the efficacy of methotrexate (MTX 25 mg/week) and mycophenolate mofetil (MMF 1.5 g twice daily). In acute VKH, MTX showed a trend toward greater reduction in foveal retinal thickness and resolution of SRD 2).
The central pathology is a CD4-positive T-cell autoimmune response against melanin proteins (tyrosinase, TRP-1, TRP-2, gp100) 3). Targets are melanocytes in the uvea (choroid), central nervous system (meninges), inner ear, and skin. Granulomatous inflammation of the choroid is the initial main lesion.
A strong genetic association with HLA-DR4 (especially DRB10405) is known 3), and linkage disequilibrium with HLA-DPB10501 has also been reported 3). About half of patients experience cold-like symptoms within two weeks before onset, suggesting that viral infections (EBV, CMV) may trigger autoimmunity through molecular mimicry.
Interpretation of imaging findings: ICG shows blurring of medium-to-large vessels and hypofluorescent spots due to choroidal circulatory disturbance. Marked early choroidal thickening on EDI-OCT reflects choroidal stromal edema. In the chronic phase, melanin loss leads to a sunset glow fundus 3).
Even with steroid pulse therapy, about 25% of cases have persistent inflammation, and gradual chorioretinal atrophy can lead to severe visual impairment in some patients. Mild subjective symptoms such as metamorphopsia and color vision abnormalities often remain.
Clinical significance of the FAST trial 2): This is the first direct comparison RCT of MTX vs. MMF in non-infectious uveitis. The 93 VKH cases were the largest subgroup, demonstrating the efficacy of early immunosuppressant introduction in acute VKH. Further follow-up studies are expected.
Importance of early immunosuppressant introduction 2): With steroid monotherapy, 44% relapse and 59% develop sunset glow fundus. Early combination of steroids with antimetabolites may reduce relapse rates and sunset glow fundus incidence. The concept of a “therapeutic window”—starting steroids early in the disease to prevent progression to chronic recurrent phase and reduce the need for long-term immunosuppression—is gaining acceptance.
Evolution of imaging diagnostics3): Quantitative assessment of choroidal thickening using EDI-OCT and detection of occult choroiditis using ICG are utilized for treatment monitoring. Quantitative measurement of hyperreflective choroidal foci (HCF) using en face OCT may become a biomarker for VKH activity evaluation.
Trends in revised diagnostic criteria2): Classification into early/late stages incorporating OCT, FA, and ICG has been proposed, and improvement in diagnostic accuracy is expected. It has been pointed out that the current 2001 revised criteria have limitations in detecting early VKH.