Phase prodromique
Période : 1 à 2 semaines avant l’apparition des symptômes oculaires
Symptômes : Sensation de picotement du cuir chevelu, céphalées (64,7 %), acouphènes (24,7 %), fatigue (21,1 %), symptômes pseudo-grippaux2)

La maladie de Vogt-Koyanagi-Harada (VKH) est une maladie auto-immune à médiation cellulaire T dirigée contre la protéine mélanique. Elle se manifeste par une uvéite, des symptômes d’irritation méningée (céphalées) et une surdité à la phase aiguë, et par des lésions cutanées telles que vitiligo, alopécie et poliosis en cas d’inflammation persistante. Des lésions inflammatoires apparaissent dans tous les tissus contenant des mélanocytes : yeux, cheveux, peau, oreille interne et méninges.
Environ la moitié des patients présentent des symptômes pseudo-grippaux dans les deux semaines précédant l’apparition de la maladie, ce qui suggère qu’une infection virale pourrait déclencher une réaction immunologique croisée (mimétisme moléculaire). Le virus d’Epstein-Barr et le cytomégalovirus sont suspectés. Le pronostic visuel est généralement bon, mais certains cas évoluent vers une déficience visuelle sévère en raison d’une inflammation persistante, d’où l’importance d’un diagnostic précoce et d’un traitement initial.
La maladie de Vogt-Koyanagi-Harada (VKH) est la deuxième cause d’uvéite. Dans une enquête épidémiologique de 2002, 205 cas de VKH (6,7 %) ont été rapportés, et en 2009, 267 cas (7,0 %), ce qui la place au deuxième rang après la sarcoïdose1). L’âge de prédilection se situe entre 20 et 50 ans, avec une légère prédominance féminine. 80 % des patients sont HLA-DR4 positifs (contre 25 % dans la population japonaise normale). La maladie est plus fréquente chez les personnes de couleur (Asiatiques, Hispaniques, Amérindiens) et rare chez les Blancs. Dans l’étude FAST, sur 93 patients VKH, 71 % étaient des femmes, avec un âge médian de 35 à 38 ans2). La maladie est la plus fréquente en Asie de l’Est, mais on la trouve également dans les régions côtières du Pacifique en Amérique du Nord et du Sud3).
La maladie est plus fréquente chez les personnes de couleur (Asiatiques, Hispaniques, Amérindiens) et rare chez les Blancs. Elle survient préférentiellement chez les personnes HLA-DR4 positives et dans la tranche d’âge 20-50 ans, avec une légère prédominance féminine. On pense qu’elle résulte d’une combinaison de prédisposition génétique (HLA-DRB1*0405) et d’un déclencheur environnemental3).
Phase prodromique
Période : 1 à 2 semaines avant l’apparition des symptômes oculaires
Symptômes : Sensation de picotement du cuir chevelu, céphalées (64,7 %), acouphènes (24,7 %), fatigue (21,1 %), symptômes pseudo-grippaux2)
Phase aiguë (phase d'apparition des symptômes oculaires)
Baisse brutale de l’acuité visuelle : Décollement séreux de la rétine bilatéral (57,6 % des VKH aigus)2)
Inflammation légère de la chambre antérieure et opacités vitréennes, rougeur et gonflement de la papille optique (environ 70 %)
Pils choroïdiens : Dans les cas sévères, décollement choroïdien et chambre antérieure peu profonde
Phase chronique (phase de récidive)
Inflammation granulomateuse du segment antérieur prédominante
KP gras, nodules de Koeppe/Busacca, synéchies postérieures de l’iris
Un fond d’œil en coucher de soleil apparaît dans 33,3 % des VKH chroniques2)
Phase tardive (récupération)
Fond d’œil en coucher de soleil (sunset glow fundus) : dépigmentation de la mélanine choroïdienne
Signe de Sugiura : dépigmentation du limbe cornéen (environ 1 mois après le début)
Symptômes cutanés (environ 20 %) : vitiligo, alopécie, poliosis (quelques mois plus tard)
Accumulation de pigment maculaire, dispersion de zones de dépigmentation chorio-rétinienne
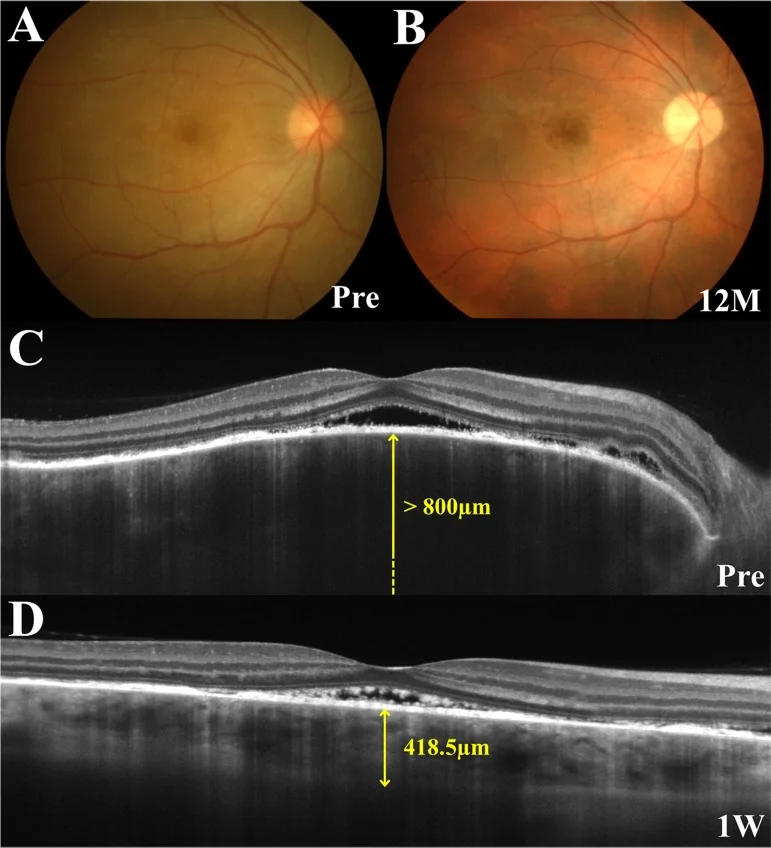
Dans la phase prodromique, des picotements du cuir chevelu, des maux de tête et des acouphènes apparaissent, suivis 1 à 2 semaines plus tard d’une baisse brutale de l’acuité visuelle et d’un flou visuel, survenant simultanément ou successivement dans les deux yeux. En OCT, la présence de structures cloisonnées à l’intérieur du décollement rétinien aigu est caractéristique.
Des symptômes d’irritation méningée (céphalées, raideur de la nuque) apparaissent précocement chez la plupart des patients. Surdité neurosensorielle (peu ressentie mais détectée à l’examen). Les symptômes cutanés (vitiligo, alopécie, poliosis) apparaissent quelques mois après le début et sont observés dans environ 20 % des cas.
Lors d’une récidive, le décollement séreux de la rétine postérieure, typique de la première poussée, est rare ; l’inflammation granulomateuse du segment antérieur prédomine. On observe des précipités rétro-cornéens (KP) gras, des nodules de Koeppe et de Busacca, et des synéchies postérieures de l’iris sont fréquentes. Des symptômes prodromiques tels qu’une rougeur oculaire et un flou visuel peuvent apparaître.
La réaction auto-immune des lymphocytes T CD4+ dirigée contre les protéines de la mélanine (famille des tyrosinases : tyrosinase, TRP-1, TRP-2, gp100) est au cœur de la pathologie 3). Les organes cibles sont les mélanocytes de l’uvée (choroïde), du système nerveux central (méninges), de l’oreille interne et de la peau ; l’inflammation granulomateuse de la choroïde constitue la lésion initiale principale.
Une forte association avec HLA-DR4 (en particulier DRB10405) est connue comme prédisposition génétique 3). Un déséquilibre de liaison avec HLA-DPB10501 a également été rapporté 3). L’hypothèse d’une infection virale (EBV, CMV) déclenchant l’auto-immunité par mimétisme moléculaire a été émise.
Principaux facteurs de risque :
| Type | Critères |
|---|---|
| Maladie de VKH complète | Signes oculaires + signes neurologiques/auditifs + manifestations cutanées, tous présents |
| Maladie de VKH incomplète | Signes oculaires présents, mais absence de signes neurologiques/auditifs ou de manifestations cutanées |
| Maladie de VKH probable | Signes oculaires uniquement (uvéite isolée) |
Au début, les lésions cutanées sont absentes, donc la plupart des cas sont incomplets. Plus tard, lorsque les lésions cutanées apparaissent, le cas devient complet. Le décollement séreux rétinien bilatéral précoce est caractéristique, et le diagnostic est facile s’il existe des symptômes prodromiques comme des céphalées ou des symptômes extraoculaires tels que des acouphènes 1). Dans les cas atypiques (œdème papillaire, unilatéral), l’examen du LCR est déterminant pour le diagnostic.
Examen du LCR : Pléiocytose à prédominance lymphocytaire. Persiste jusqu’à 8 semaines. Le plus utile pour confirmer le diagnostic.
Angiographie à la fluorescéine (FA) : Hypofluorescence en plaques due à un retard de remplissage choroïdien précoce → fuites ponctuelles (pinpoint leakage) à la phase intermédiaire → accumulation de colorant dans la zone de SRD à la phase tardive. Hyperfluorescence de la papille optique (environ 70 %) 1).
Angiographie au vert d’indocyanine (ICG) : Retard de remplissage choroïdien en plaques et fuite de colorant des vaisseaux choroïdiens à la phase précoce, taches hypofluorescentes disséminées aux phases intermédiaire et tardive. L’effacement des vaisseaux moyens et grands dû à un trouble circulatoire choroïdien est également caractéristique.
OCT : Détection et suivi du décollement séreux rétinien. Épaississement choroïdien marqué au début. L’EDI-OCT permet d’observer en détail la coupe transversale de la choroïde. Des structures membranaires fibrineuses et des septa peuvent être observés dans le SRD 1).
Échographie en mode B : Épaississement choroïdien. Utile pour le diagnostic différentiel avec la sclérite postérieure 1).
HLA de classe II : HLA-DR4 (examen complémentaire). Positif dans 80 % des cas, mais spécificité faible (25 % de positifs chez les sujets sains).
ERG (électrorétinogramme) : Diminution des amplitudes des réponses photopiques et scotopiques en phase chronique 3).
| Maladie différentielle | Points de différenciation |
|---|---|
| Sclérite postérieure | Unilatéral, signe du T à l’échographie, douleur aux mouvements oculaires |
| Épithéliopathie pigmentaire placoïde postérieure multifocale (MPPE) | Unilatéral, décollement séreux rétinien mobile et bulleux |
| Épithéliopathie pigmentaire rétinienne aiguë postérieure multifocale (APMPPE) | Phénomène d’inversion à l’angiographie à la fluorescéine (hyperfluorescence précoce → hypofluorescence tardive), sujet jeune, infection antérieure |
| Choriorétinopathie séreuse centrale (CRSC) | Unilatéral, prédominance masculine, les stéroïdes sont un facteur aggravant3) |
| Ophtalmie sympathique | Différenciation par antécédent de plaie perforante oculaire ou de chirurgie intraoculaire. Considérée pathologiquement comme la même maladie1) |
| Syndrome d’exsudation uvéale idiopathique | Association avec la nanophtalmie, attention à la fermeture de l’angle |
Elle n’est pas indispensable, mais elle est déterminante dans les cas atypiques difficiles à diagnostiquer (type œdème papillaire, cas unilatéral). La pléiocytose à prédominance lymphocytaire persiste jusqu’à 8 semaines, donc la réaliser au moment approprié après le début améliore la précision diagnostique. Dans les cas typiques avec décollement séreux rétinien bilatéral évident, le diagnostic peut souvent être posé sur la seule base des signes cliniques.
La corticothérapie par bolus suivie d’une dégression orale à forte dose est le traitement standard. En l’absence de traitement, de nombreux cas évoluent vers des récidives ou une chronicité, entraînant une déficience visuelle sévère. Comme il n’existe actuellement aucun indicateur permettant de distinguer les cas de guérison spontanée des cas récidivants ou chroniques, un traitement par corticostéroïdes à forte dose est recommandé dans la mesure du possible.
Exemple de prescription représentative des directives1) :
Calendrier de diminution progressive orale (Prednisone comprimés 5 mg) :
| Dose | Durée |
|---|---|
| 200 mg/jour | 2 jours |
| 150 mg/jour | 2 jours |
| 100 mg/jour | 2 jours |
| 80 mg/jour | 2 jours |
| 60 mg/jour | 4 jours |
| 40 mg/jour | 10 jours |
| 30 mg/jour | 2 semaines |
| 20 mg/jour | 4 semaines |
| 15 mg/jour | 4 semaines |
| 10 mg/jour | 4 semaines |
| 5 mg/jour | 4 semaines |
| 5 mg/jour (un jour sur deux) | 4 semaines |
La réduction doit être lente, et le traitement doit être arrêté sur une période d’au moins 6 mois même en l’absence de récidive. Avec un traitement de moins de 6 mois, le taux de récidive est de 58,8 %, contre 11,1 % pour un traitement de 6 mois ou plus 5). En cas de récidive, augmenter la dose de stéroïdes et réduire progressivement sur une période plus longue que la précédente. Avec la monothérapie stéroïdienne, 44 % des patients présentent une récidive et 59 % développent un fundus en coucher de soleil 2).
Doses pour les patients pédiatriques : Chez les enfants d’un âge et d’un poids suffisants, une perfusion de méthylprednisolone à 500 mg/jour pendant 3 jours (thérapie par impulsions) ou une réduction progressive de prednisolone par voie orale à partir de 0,5 à 1,0 mg/kg/jour est réalisée. La réduction se fait en surveillant les effets secondaires.
Ciclosporine (Néoral®) : 3 mg/kg/jour (pour un poids de 60 kg, 180 mg/jour en deux prises). Une surveillance régulière de la concentration résiduelle est nécessaire. Permet de réduire la dose de stéroïdes. Applicable aux cas prolongés 1). Attention aux effets secondaires tels que sensibilité aux infections, insuffisance rénale et hépatique.
Méthotrexate (MTX) : 25 mg/semaine par voie orale. Commencer à 15 mg/semaine pendant 2 semaines, puis augmenter 2).
Mycophénolate mofétil (MMF) : 1,5 g deux fois par jour par voie orale. Commencer à 500 mg deux fois par jour et augmenter progressivement 2).
L’essai FAST (NCT01829295) est un essai randomisé contrôlé sur les uvéites non infectieuses (sous-analyse de 93 cas de VKH sur 216 patients). 49 patients ont été randomisés dans le groupe MTX et 44 dans le groupe MMF, avec prednisone à 1 mg/kg/jour (max 60 mg/jour) en débutant puis réduction progressive (objectif ≤ 7,5 mg/jour à 6 mois).
Critère principal à 6 mois (sous-analyse VKH) :
| Indicateur | Groupe MTX | Groupe MMF | Valeur P |
|---|---|---|---|
| Taux de succès thérapeutique | 80,4 % (37/46 cas) | 64,1 % (25/39 cas) | 0,10 |
| Réduction de l’épaisseur rétinienne fovéale | −62,5 μm | −4,0 μm | 0,003 |
| Taux de disparition du SRD | 86,3 % | 64,1 % | 0,02 |
| Amélioration de la vision | Équivalent | Équivalent | 0,78 |
Le taux de succès thérapeutique global était de 74,7 % (62/85 cas). Dans la VKH aiguë, le MTX était supérieur pour la réduction de l’épaisseur centrale de la rétine (CST) et la disparition du décollement séreux rétinien (SRD). À 12 mois, 91,3 % du groupe MMF maintenaient le succès thérapeutique, et environ la moitié (MTX 50,0 %, MMF 56,5 %) avaient atteint un sevrage complet de la prednisone 2). Le passage de MMF à MTX après un échec du premier traitement a réussi dans 81,8 % des cas 2). L’utilisation initiale de MMF en association a permis à 93 % des VKH aiguës de maintenir une acuité visuelle de 20/20, sans récidive ni formation de fundus en coucher de soleil chez tous les patients 2).
Chez les patients pour lesquels l’administration systémique de stéroïdes est difficile (personnes âgées, femmes enceintes, diabétiques, antécédents de maladie mentale), envisager une injection sous-ténonienne postérieure de triamcinolone acétonide 1).
Cataracte concomitante : L’utilisation intensive de stéroïdes entraîne une incidence élevée de cataracte stéroïdienne. Les cas en rémission complète présentent un risque similaire à celui d’une chirurgie standard. L’implantation d’un cristallin artificiel (IOL) ne pose pas de problème. Si une chirurgie filtrante est ultérieurement nécessaire, choisir une incision cornéenne en préservant la conjonctive supérieure.
Glaucome secondaire / glaucome stéroïdien : Utiliser des collyres hypotenseurs (bêta-bloquants, analogues des prostaglandines, inhibiteurs de l’anhydrase carbonique) → inhibiteurs de l’anhydrase carbonique par voie orale → perfusion de D-mannitol dans cet ordre. Effectuer une trabéculotomie (efficace pour le glaucome stéroïdien), et si insuffisante, une trabéculectomie.
Même en l’absence de récidive, il est recommandé de réduire progressivement la dose sur au moins 6 mois. L’arrêt en moins de 6 mois entraîne un taux de récidive élevé d’environ 58,8 %, tandis que la poursuite au-delà de 6 mois réduit ce taux à 11,1 % 5). En cas de récidive, réduire la dose plus lentement que la fois précédente et envisager l’ajout d’un immunosuppresseur.
La ciclosporine (Neoral® 3 mg/kg/jour) est utilisée comme épargneur de stéroïdes dans les cas persistants 1). L’essai FAST a confirmé l’efficacité du méthotrexate (MTX 25 mg/semaine) et du mycophénolate mofétil (MMF 1,5 g × 2 fois/jour). Dans la VKH aiguë, le MTX a montré une tendance supérieure dans la réduction de l’épaisseur rétinienne fovéale et le taux de disparition du décollement séreux rétinien 2).
La réaction auto-immune à médiation cellulaire T CD4+ dirigée contre les protéines de la mélanine (tyrosinase, TRP-1, TRP-2, gp100) est le mécanisme central 3). Les cibles sont les mélanocytes de l’uvée (choroïde), du système nerveux central (méninges), de l’oreille interne et de la peau ; l’inflammation granulomateuse de la choroïde constitue la lésion initiale principale.
Une forte association avec HLA-DR4 (en particulier DRB10405) est connue comme prédisposition génétique 3), et un déséquilibre de liaison avec HLA-DPB10501 a également été rapporté 3). Environ la moitié des patients présentent des symptômes pseudo-grippaux dans les deux semaines précédant l’apparition, ce qui suggère qu’une infection virale (EBV, CMV) pourrait déclencher l’auto-immunité par mimétisme moléculaire.
Interprétation des résultats d’imagerie : L’ICG montre un flou des vaisseaux moyens et grands et des taches hypofluorescentes dus à des troubles circulatoires choroïdiens. L’épaississement choroïdien marqué précocement à l’EDI-OCT reflète un œdème du stroma choroïdien. La dépigmentation mélanique en phase chronique conduit à un fond d’œil en coucher de soleil 3).
Environ 25 % des cas présentent une inflammation persistante même après un traitement par bolus de stéroïdes ; la persistance de l’inflammation choroïdienne entraîne une atrophie choroïdienne et rétinienne progressive, conduisant à une déficience visuelle sévère chez certains patients. Des symptômes subjectifs tels qu’une légère métamorphopsie ou des anomalies de la vision des couleurs persistent souvent.
Signification clinique de l’essai FAST 2) : Premier essai contrôlé randomisé comparant directement MTX et MMF dans l’uvéite non infectieuse. Les 93 cas de VKH constituaient le plus grand sous-groupe de l’essai, démontrant l’efficacité de l’introduction précoce d’immunosuppresseurs dans la VKH aiguë. Des études de suivi supplémentaires sont attendues.
Importance de l’introduction précoce d’immunosuppresseurs 2) : Avec la corticothérapie seule, 44 % des patients présentent une récidive et 59 % développent un fond d’œil en coucher de soleil. L’association précoce de stéroïdes et d’antimétabolites peut réduire les taux de récidive et d’apparition du fond d’œil en coucher de soleil. La notion de « fenêtre thérapeutique » — l’instauration précoce de stéroïdes au début de la maladie prévient la progression vers la phase chronique récurrente et réduit le besoin d’immunosuppression à long terme — gagne du terrain.
Évolution du diagnostic par imagerie3) : L’évaluation quantitative de l’épaisseur choroïdienne par EDI-OCT et la détection de la choriorétinite occultée par ICG sont utilisées pour le suivi thérapeutique. La mesure quantitative des foyers choroïdiens hyperréflectifs (HCF) par en face OCT pourrait constituer un biomarqueur de l’activité de la maladie de VKH.
Évolution des critères diagnostiques2) : Une classification précoce/tardive intégrant l’OCT, l’angiographie à la fluorescéine (FA) et l’ICG a été proposée, ce qui devrait améliorer la précision diagnostique. Les critères révisés de 2001 présentent des limites pour la détection précoce de la maladie de VKH.