مرحله پیشدرآمدی
زمان: 1 تا 2 هفته قبل از بروز علائم چشمی
علائم: احساس سوزنسوزن شدن موی سر، سردرد (64.7%)، وزوز گوش (24.7%)، خستگی (21.1%)، علائم شبه سرماخوردگی2)

بیماری فوگت-کویاناگی-هارادا (Vogt-Koyanagi-Harada disease; VKH) یک بیماری خودایمنی سلول T علیه پروتئین ملانین است. در مرحله شروع، یووئیت، علائم تحریک مننژ مانند سردرد و کاهش شنوایی را نشان میدهد و با تداوم التهاب، علائم پوستی مانند لکههای سفید، ریزش مو و سفید شدن مو بروز میکند. این یک بیماری سیستمیک است که در تمام نواحی دارای ملانوسیتها - چشم، مو، پوست، گوش داخلی و مننژ - ضایعات التهابی ایجاد میکند.
از آنجایی که حدود نیمی از بیماران در عرض 2 هفته قبل از شروع علائم شبه سرماخوردگی دارند، تصور میشود عفونت ویروسی به عنوان محرک از طریق تقلید مولکولی (molecular mimicry) باعث شروع بیماری میشود. نقش ویروس اپشتین-بار و سیتومگالوویروس مطرح شده است. به طور کلی پیشآگهی بینایی خوب است، اما در برخی موارد تداوم التهاب منجر به اختلال شدید بینایی میشود، بنابراین تشخیص زودهنگام و درمان اولیه مهم است.
بیماری VKH دومین علت شایع یووئیت است. در بررسی اپیدمیولوژیک سال 2002، 205 مورد (6.7%) و در سال 2009، 267 مورد (7.0%) گزارش شده است که در هر دو، پس از سارکوئیدوز در رتبه دوم قرار دارد1). سن شایع بروز 20 تا 50 سالگی است و در زنان کمی شایعتر است. 80% بیماران HLA-DR4 مثبت هستند (در حالی که 25% افراد عادی ژاپنی HLA-DR4 مثبت هستند). این بیماری در نژادهای رنگی (آسیاییها، اسپانیاییتبارها، بومیان آمریکا) شایعتر و در سفیدپوستان نادر است. از 93 بیمار VKH ثبتشده در مطالعه FAST، 71% زن و میانه سنی 35-38 سال بود2). این بیماری در شرق آسیا شایعترین است، اما در مناطق ساحلی اقیانوس آرام در آمریکای شمالی و جنوبی نیز دیده میشود3).
این بیماری در نژادهای رنگی (آسیاییها، اسپانیاییتبارها، بومیان آمریکا) شایعتر و در سفیدپوستان نادر است. در افراد HLA-DR4 مثبت و در سنین 20-50 سالگی با غلبه نسبی در زنان شایع است. تصور میشود که ترکیبی از استعداد ژنتیکی (HLA-DRB1*0405) و یک محرک محیطی باعث بروز بیماری میشود3).
مرحله پیشدرآمدی
زمان: 1 تا 2 هفته قبل از بروز علائم چشمی
علائم: احساس سوزنسوزن شدن موی سر، سردرد (64.7%)، وزوز گوش (24.7%)، خستگی (21.1%)، علائم شبه سرماخوردگی2)
مرحله حاد (مرحله بروز علائم چشمی)
کاهش شدید بینایی: جداشدگی سروزی شبکیه دوطرفه (57.6% در VKH حاد)2)
التهاب خفیف اتاق قدامی و کدورت زجاجیه، قرمزی و تورم دیسک بینایی (حدود 70%)
چینهای مشیمیه: در موارد شدید، جداشدگی مشیمیه و اتاق قدامی کم عمق
مرحله مزمن (مرحله عود)
التهاب گرانولوماتوز بخش قدامی چشم غالب است
رسوبات چربیمانند روی قرنیه (KP)، ندولهای Koeppe/Busacca، چسبندگی عنبیه به عدسی
در 33.3% از موارد VKH مزمن، فوندوس غروبآفتابی ظاهر میشود2)
مرحله دیررس (بهبودی)
فوندوس غروبآفتابی (sunset glow fundus): از دست رفتن رنگدانه ملانین مشیمیه
علامت سوگیورا: از دست رفتن رنگدانه در لبه قرنیه (حدود یک ماه پس از شروع)
علائم پوستی (حدود 20%): لکهای سفید، ریزش مو، سفید شدن مو (چند ماه بعد)
تجمع رنگدانه در ناحیه ماکولا، لکههای پراکنده از دست رفتن رنگدانه شبکیه و مشیمیه
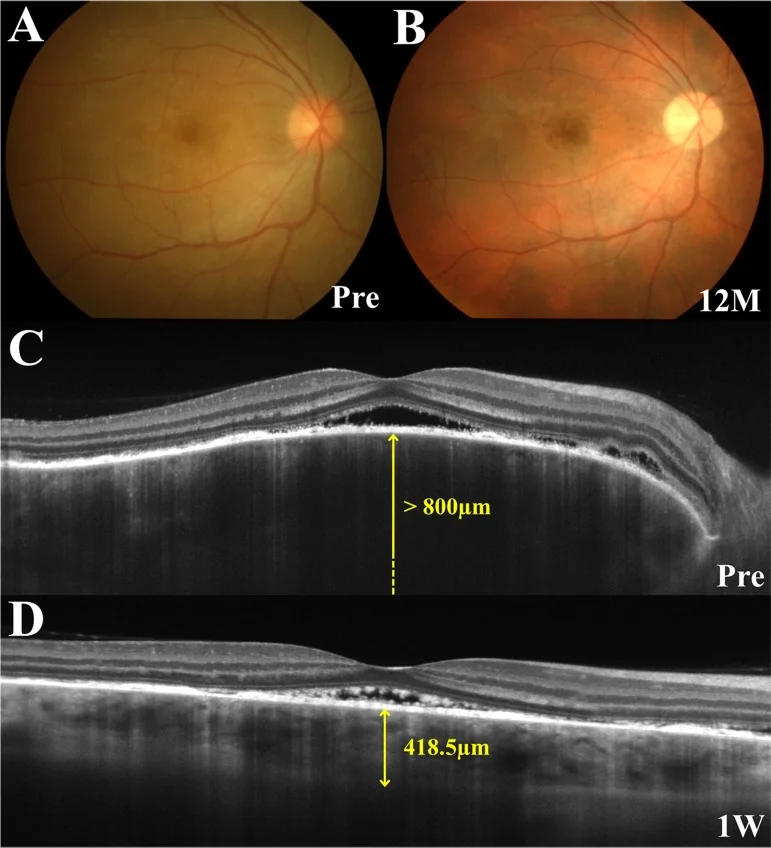
در مرحله پیشدرآمدی، احساس سوزن سوزن شدن موی سر، سردرد و وزوز گوش ظاهر میشود و یک تا دو هفته بعد، کاهش شدید بینایی و تار شدن دید به طور همزمان در هر دو چشم یا با فاصله رخ میدهد. در OCT، در مرحله حاد، ساختارهای سپتوممانند در داخل جداشدگی شبکیه مشاهده میشود که مشخصه است.
علائم تحریک مننژ (سردرد، سفتی گردن) در اکثر موارد در اوایل بیماری ظاهر میشود. کاهش شنوایی حسی-عصبی (که اغلب توسط بیمار احساس نمیشود اما در آزمایش مشخص میگردد). علائم پوستی (لکهای سفید، ریزش مو، سفید شدن مو) چند ماه پس از شروع بیماری ظاهر میشود و در حدود 20% موارد دیده میشود.
در زمان عود، جداشدگی سروزی شبکیه خلفی مانند مرحله اولیه کمتر دیده میشود و التهاب گرانولوماتوز بخش قدامی چشم غالب است. رسوبات چربیمانند روی قرنیه (KP)، ندولهای Koeppe و Busacca مشاهده میشود و اغلب چسبندگی عنبیه به عدسی رخ میدهد. علائم پیشدرآمدی ممکن است شامل قرمزی چشم و تاری دید باشد.
واکنش خودایمنی سلولهای T CD4+ علیه پروتئین ملانین (خانواده تیروزیناز: تیروزیناز، TRP-1، TRP-2، gp100) هسته اصلی پاتوژنز است 3). اندامهای هدف شامل ملانوسیتهای یووه آ (مشیمیه)، سیستم عصبی مرکزی (مننژ)، گوش داخلی و پوست هستند و التهاب گرانولوماتوز مشیمیه ضایعه اولیه اصلی است.
استعداد ژنتیکی با HLA-DR4 (به ویژه DRB10405) ارتباط قوی دارد 3). عدم تعادل پیوندی با HLA-DPB10501 نیز گزارش شده است 3). فرضیهای وجود دارد که عفونتهای ویروسی (EBV، CMV) از طریق تقلید مولکولی باعث ایجاد خودایمنی میشوند.
عوامل خطر اصلی:
| نوع | معیار |
|---|---|
| بیماری کامل VKH (Complete VKH disease) | یافتههای چشمی + یافتههای عصبی/شنوایی + علائم پوستی، همه را شامل میشود |
| بیماری ناقص VKH (Incomplete VKH disease) | یافتههای چشمی وجود دارد، اما یکی از یافتههای عصبی/شنوایی یا علائم پوستی وجود ندارد |
| بیماری مشکوک VKH (Probable VKH disease) | فقط یافتههای چشمی (یووئیت ایزوله) |
در مراحل اولیه بیماری، به دلیل عدم وجود علائم پوستی، بیشتر موارد از نوع ناقص هستند. بعداً با ظهور علائم پوستی، به نوع کامل تبدیل میشوند. جداشدگی سروز دوطرفه شبکیه در مراحل اولیه مشخصه است و در صورت وجود علائم پیشدرآمدی مانند سردرد یا علائم خارج چشمی مانند وزوز گوش، تشخیص آسان است 1). در موارد غیر معمول (نوع ادم پاپی عصب بینایی یا موارد یک طرفه)، آزمایش مایع مغزی-نخاعی عامل تعیینکننده تشخیص است.
آزمایش مایع مغزی-نخاعی: افزایش سلولها با غلبه لنفوسیتها. تا ۸ هفته ادامه دارد. مفیدترین روش برای تأیید تشخیص.
آنژیوگرافی فلورسین (FA): در مراحل اولیه، تأخیر در پر شدن مشیمیه به صورت لکههای هیپوفلورسنت → در مرحله میانی، نشت نقطهای فلورسین (pinpoint leakage) → در مرحله پایانی، تجمع رنگ در ناحیه جداشدگی سروز شبکیه. هیپرفلورسنس پاپی عصب بینایی (حدود ۷۰٪) 1).
آنژیوگرافی با ایندوسیانین گرین (ICG): در مراحل اولیه، تأخیر در پر شدن لکهای مشیمیه و نشت رنگ از عروق مشیمیه، در مراحل میانی تا پایانی، لکههای هیپوفلورسنت پراکنده. تار شدن عروق متوسط و بزرگ به دلیل اختلال گردش خون مشیمیه نیز مشخصه است.
OCT: تشخیص و پایش جداشدگی سروز شبکیه. در مراحل اولیه، ضخیم شدن قابل توجه مشیمیه. با EDI-OCT میتوان مقطع مشیمیه را با جزئیات مشاهده کرد. ممکن است ساختارهای غشایی فیبرینی و سپتومها در داخل جداشدگی سروز شبکیه دیده شود 1).
سونوگرافی B-mode: یافتههای ضخیم شدن مشیمیه. برای افتراق از اسکلریت خلفی مفید است 1).
HLA class II: HLA-DR4 (آزمایش کمکی). میزان مثبت شدن ۸۰٪ اما اختصاصیت پایین است (در افراد سالم نیز ۲۵٪ مثبت).
ERG (الکترورتینوگرام): در مرحله مزمن، دامنه امواج روشنایی و تطابق با تاریکی کاهش مییابد 3).
| بیماری افتراقی | نکات افتراقی |
|---|---|
| اسکلریت خلفی | یک طرفه، علامت T در سونوگرافی، درد با حرکت چشم و حساسیت |
| اپیتلیوپاتی رنگدانهای چندکانونی خلفی (MPPE) | یک طرفه، جداشدگی سروز تاولی مهاجر |
| اپیتلیوپاتی حاد پلاسید چندکانونی خلفی رنگدانهای (APMPPE) | پدیده معکوس FA (هایپرفلورسانس اولیه → هیپوفلورسانس دیررس)، سن جوان، عفونت قبلی |
| کوریورتینوپاتی سروز مرکزی (CSCR) | یک طرفه، غالب در مردان، استروئیدها عامل تشدید کننده 3) |
| افتالمی سمپاتیک | تشخیص افتراقی با سابقه ترومای نافذ چشمی یا جراحی داخل چشمی. از نظر پاتولوژیک همان بیماری در نظر گرفته میشود 1) |
| سندرم افیوژن یووهآی ایدیوپاتیک | ارتباط با نانوفتالموس، توجه به بسته شدن زاویه |
ضروری نیست، اما در موارد غیر معمول که تشخیص دشوار است (نوع ادم پاپی، موارد یک طرفه) تعیین کننده است. از آنجایی که پلئوسیتوز با سلولهای غالب لنفوسیتی تا ۸ هفته ادامه دارد، انجام آن در زمان مناسب پس از شروع بیماری دقت تشخیص را افزایش میدهد. در موارد معمول با جداشدگی سروز دو طرفه واضح شبکیه، اغلب میتوان تنها بر اساس یافتههای بالینی تشخیص داد.
پالس درمانی استروئیدی → کاهش تدریجی خوراکی با دوز بالا استاندارد است. در صورت عدم درمان و مشاهده، بسیاری از موارد دچار عود و مزمن شدن شده و به اختلال شدید بینایی منجر میشوند. از آنجایی که در پزشکی فعلی شاخصی برای تمایز موارد بهبودی خودبهخودی از موارد عود و مزمن وجود ندارد، در صورت امکان تجویز دوز بالای استروئید توصیه میشود.
نمونه نسخه نماینده راهنما1):
برنامه کاهش تدریجی خوراکی (قرص پردنین 5 میلیگرم):
| دوز | مدت |
|---|---|
| 200 میلیگرم/روز | 2 روز |
| 150 میلیگرم/روز | 2 روز |
| 100 میلیگرم/روز | 2 روز |
| 80 میلیگرم/روز | 2 روز |
| 60 میلیگرم در روز | 4 روز |
| 40 میلیگرم در روز | 10 روز |
| 30 میلیگرم در روز | 2 هفته |
| 20 میلیگرم در روز | 4 هفته |
| 15 میلیگرم در روز | 4 هفته |
| 10 میلیگرم در روز | 4 هفته |
| 5 میلیگرم در روز | 4 هفته |
| 5 میلیگرم در روز (یک روز در میان) | 4 هفته |
کاهش دوز به آرامی انجام میشود و حتی در صورت عدم عود، طی ۶ ماه یا بیشتر قطع میشود. با درمان کمتر از ۶ ماه، میزان عود ۵۸.۸٪ و با درمان ۶ ماه یا بیشتر به ۱۱.۱٪ کاهش مییابد5). در صورت عود، دوز استروئید افزایش یافته و نسبت به دفعه قبل با زمان بیشتری کاهش مییابد. با درمان تکدارویی استروئید، ۴۴٪ عود و در ۵۹٪ فوندوس sunset glow ایجاد میشود2).
دوز درمانی در کودکان: در کودکان با سن و وزن مشخص، متیلپردنیزولون ۵۰۰ میلیگرم در روز به صورت انفوزیون وریدی به مدت ۳ روز (پالس درمانی) یا پردنیزولون ۰.۵ تا ۱.۰ میلیگرم/کیلوگرم/روز به صورت خوراکی با کاهش تدریجی تجویز میشود. کاهش دوز با توجه به عوارض جانبی انجام میشود.
سیکلوسپورین (Neoral®): ۳ میلیگرم/کیلوگرم/روز (برای وزن ۶۰ کیلوگرم: ۱۸۰ میلیگرم/روز در دو دوز منقسم). نیاز به اندازهگیری منظم سطح خونی (تراف) دارد. انتظار میرود دوز استروئید کاهش یابد. در موارد مقاوم استفاده میشود1). مراقب عوارضی مانند افزایش حساسیت به عفونت، اختلال عملکرد کلیه و کبد باشید.
متوترکسات (MTX): ۲۵ میلیگرم در هفته خوراکی. شروع با ۱۵ میلیگرم در هفته به مدت ۲ هفته و سپس افزایش دوز2).
میکوفنولات موفتیل (MMF): ۱.۵ گرم دو بار در روز خوراکی. شروع با ۵۰۰ میلیگرم دو بار در روز و سپس افزایش تدریجی2).
آزمایش FAST (NCT01829295) یک کارآزمایی بالینی تصادفیشده بر روی یووئیت غیرعفونی (از ۲۱۶ بیمار، ۹۳ مورد VKH در آنالیز زیرگروه) است. ۴۹ بیمار MTX و ۴۴ بیمار MMF دریافت کردند و با پردنیزون ۱ میلیگرم/کیلوگرم/روز (حداکثر ۶۰ میلیگرم/روز) شروع و سپس کاهش یافت (هدف ≤۷.۵ میلیگرم/روز در ۶ ماه).
پیامد اولیه ۶ ماهه (تحلیل زیرگروه VKH):
| شاخص | گروه MTX | گروه MMF | مقدار P |
|---|---|---|---|
| نرخ موفقیت درمان | ۸۰.۴٪ (۳۷/۴۶ مورد) | ۶۴.۱٪ (۲۵/۳۹ مورد) | ۰.۱۰ |
| کاهش ضخامت شبکیه فووئال | ۶۲.۵- میکرومتر | ۴.۰- میکرومتر | ۰.۰۰۳ |
| نرخ ناپدید شدن SRD | ۸۶.۳٪ | ۶۴.۱٪ | ۰.۰۲ |
| بهبود بینایی | معادل | معادل | 0.78 |
نرخ موفقیت کلی درمان 74.7% (62 از 85 بیمار) بود. در VKH حاد، MTX از نظر کاهش CST و ناپدید شدن SRD برتر بود. در ماه 12، 91.3% از گروه MMF به درمان موفق ادامه دادند و حدود نیمی (MTX 50.0%، MMF 56.5%) به قطع کامل پردنیزون دست یافتند2). در تغییر از MMF به MTX پس از شکست درمان اولیه، 81.8% موفق بودند2). گزارش شده است که با مصرف اولیه MMF، 93% از موارد VKH حاد دید 20/20 را حفظ کردند و هیچ عود یا تشکیل sunset glow fundus در هیچیک از موارد مشاهده نشد2).
در مواردی که تجویز سیستمیک استروئید دشوار است (سالمندان، زنان باردار، دیابت، سابقه بیماری روانی)، تزریق زیر تانون خلفی تریامسینولون استونید را در نظر بگیرید1).
آب مروارید همزمان: استفاده زیاد از استروئیدها منجر به بروز بالای آب مروارید استروئیدی میشود. موارد بهبودی کامل خطر مشابه جراحی معمولی دارند. کاشت IOL نیز مشکلی ندارد. در صورت احتمال جراحی فیلتراسیون بعدی، ملتحمه فوقانی را حفظ کرده و برش قرنیه را انتخاب کنید.
گلوکوم ثانویه و گلوکوم استروئیدی: از قطرههای کاهنده فشار (مسدودکنندههای بتا، آنالوگهای پروستاگلاندین، مهارکنندههای کربنیک آنهیدراز) → سپس CAI خوراکی → سپس انفوزیون D-مانیتول استفاده کنید. ترابکولوتومی (برای گلوکوم استروئیدی مؤثر است) و در صورت ناکافی بودن، ترابکولکتومی انجام دهید.
حتی بدون عود، توصیه میشود که به آرامی طی 6 ماه یا بیشتر کاهش دوز داده شود. قطع درمان در کمتر از 6 ماه با نرخ عود حدود 58.8% همراه است، در حالی که ادامه بیش از 6 ماه آن را به 11.1% کاهش میدهد5). در صورت عود، دوز را در مدت زمان طولانیتری نسبت به قبل کاهش دهید و افزودن داروی سرکوبکننده ایمنی را نیز در نظر بگیرید.
سیکلوسپورین (Neoral® 3 mg/kg/day) به عنوان داروی کاهشدهنده استروئید در موارد مقاوم استفاده میشود1). در مطالعه FAST، اثربخشی متوترکسات (MTX 25 mg/week) و مایکوفنولات موفتیل (MMF 1.5 g دو بار در روز) تأیید شد. در VKH حاد، MTX تمایل به برتری در کاهش ضخامت ماکولا و نرخ ناپدید شدن SRD نشان داد2).
واکنش خودایمنی سلول T CD4+ علیه پروتئینهای ملانین (تیروزیناز، TRP-1، TRP-2، gp100) مکانیسم اصلی بیماری است3). هدف، ملانوسیتهای یووه (مشیمیه)، سیستم عصبی مرکزی (مننژ)، گوش داخلی و پوست هستند و التهاب گرانولوماتوز مشیمیه ضایعه اولیه اصلی است.
از نظر استعداد ژنتیکی، ارتباط قوی با HLA-DR4 (به ویژه DRB10405) شناخته شده است3) و عدم تعادل پیوندی با HLA-DPB10501 نیز گزارش شده است3). از آنجایی که حدود نیمی از بیماران در عرض دو هفته قبل از شروع علائم شبه سرماخوردگی دارند، این فرضیه وجود دارد که عفونت ویروسی (EBV، CMV) از طریق تقلید مولکولی خودایمنی را القا میکند.
تفسیر یافتههای تصویربرداری: در ICG، تار شدن عروق متوسط و بزرگ و لکههای هیپوفلورسنت ناشی از اختلال گردش خون مشیمیه مشاهده میشود. ضخیم شدن قابل توجه اولیه مشیمیه در EDI-OCT نشاندهنده ادم بینابینی مشیمیه است. در مرحله مزمن، دپیگمانتاسیون ملانین منجر به فوندوس غروب آفتاب میشود3).
حتی با پالس درمانی استروئیدی، حدود 25٪ موارد التهاب مقاوم دارند و در برخی بیماران، التهاب مداوم در مشیمیه به تدریج باعث آتروفی گسترده شبکیه-مشیمیه و اختلال شدید بینایی میشود. علائم ذهنی مانند دیستورشن خفیف یا اختلال دید رنگی اغلب باقی میماند.
اهمیت بالینی مطالعه FAST2): اولین RCT مقایسه مستقیم MTX در مقابل MMF در یووئیت غیرعفونی. 93 مورد VKH بزرگترین زیرگروه در مطالعه بودند و اثربخشی معرفی زودهنگام داروی سرکوبکننده ایمنی در VKH حاد نشان داده شد. تحقیقات پیگیری بیشتر در آینده مورد انتظار است.
اهمیت معرفی زودهنگام داروی سرکوبکننده ایمنی2): در درمان با استروئید به تنهایی، 44٪ عود و 59٪ فوندوس غروب آفتاب ایجاد میشود. ترکیب زودهنگام استروئید + آنتیمتابولیت ممکن است نرخ عود و بروز فوندوس غروب آفتاب را کاهش دهد. «پنجره درمانی» - این ایده که شروع زودهنگام استروئید در مراحل اولیه بیماری از پیشرفت به مرحله عود مزمن جلوگیری کرده و نیاز به درمان سرکوبکننده ایمنی طولانیمدت را کاهش میدهد، در حال گسترش است.
پیشرفت در تصویربرداری تشخیصی3): ارزیابی کمی ضخامت مشیمیه با EDI-OCT و تشخیص کوروئیدیت نهفته با ICG در پایش درمان استفاده میشود. اندازهگیری کمی کانونهای هایپررفلکتیو مشیمیه (HCF) با en face OCT ممکن است به عنوان نشانگر زیستی برای ارزیابی فعالیت VKH عمل کند.
روندهای تجدید نظر در معیارهای تشخیصی2): طبقهبندی زودرس/دیررس با استفاده از OCT، FA و ICG پیشنهاد شده است که انتظار میرود دقت تشخیصی را بهبود بخشد. اشاره شده است که معیارهای تجدید نظر شده فعلی (2001) در تشخیص VKH زودرس محدودیت دارند.