Prodromalphase
Zeitraum : 1–2 Wochen vor Auftreten der Augensymptome
Symptome : Kribbelgefühl auf der Kopfhaut, Kopfschmerzen (64,7 %), Tinnitus (24,7 %), Müdigkeit (21,1 %), grippeähnliche Symptome2)

Morbus Vogt-Koyanagi-Harada (VKH) ist eine T-Zell-vermittelte Autoimmunerkrankung gegen Melaninproteine. In der akuten Phase treten Uveitis, meningeale Reizsymptome wie Kopfschmerzen und Hörverlust auf; bei anhaltender Entzündung entwickeln sich Hautveränderungen wie Vitiligo, Alopezie und Poliosis. Entzündliche Läsionen treten in allen melanosythaltigen Geweben auf – Augen, Haare, Haut, Innenohr und Meningen.
Etwa die Hälfte der Patienten zeigt innerhalb von zwei Wochen vor Krankheitsbeginn grippeähnliche Symptome, was vermuten lässt, dass eine Virusinfektion als Auslöser dient und die Erkrankung über eine immunologische Kreuzreaktion (molekulares Mimikry) entsteht. Epstein-Barr-Virus und Zytomegalievirus werden als mögliche Auslöser diskutiert. Die Sehprognose ist im Allgemeinen gut, aber in einigen Fällen führt die anhaltende Entzündung zu schwerer Sehbehinderung, weshalb eine frühzeitige Diagnose und Behandlung wichtig sind.
Die Vogt-Koyanagi-Harada-Krankheit (VKH) ist die zweithäufigste Ursache einer Uveitis. In einer epidemiologischen Erhebung von 2002 wurden 205 VKH-Fälle (6,7 %) und 2009 267 Fälle (7,0 %) berichtet, jeweils an zweiter Stelle nach der Sarkoidose1). Das bevorzugte Alter liegt zwischen 20 und 50 Jahren, mit einer leichten weiblichen Prädominanz. 80 % der Patienten sind HLA-DR4-positiv (25 % der normalen japanischen Bevölkerung sind HLA-DR4-positiv). Die Erkrankung tritt häufiger bei farbigen Menschen (Asiaten, Hispanics, Ureinwohner Amerikas) auf und ist bei Weißen selten. In der FAST-Studie waren von 93 VKH-Patienten 71 % weiblich, das mediane Alter betrug 35–38 Jahre2). Am häufigsten ist sie in Ostasien, kommt aber auch in den pazifischen Küstenregionen Nord- und Südamerikas vor3).
Die Erkrankung tritt häufiger bei farbigen Menschen (Asiaten, Hispanics, Ureinwohner Amerikas) auf und ist bei Weißen selten. Sie tritt bevorzugt bei HLA-DR4-positiven Personen und in der Altersgruppe 20–50 Jahre mit einer leichten weiblichen Prädominanz auf. Es wird angenommen, dass sie durch eine Kombination aus genetischer Veranlagung (HLA-DRB1*0405) und einem Umweltauslöser entsteht3).
Prodromalphase
Zeitraum : 1–2 Wochen vor Auftreten der Augensymptome
Symptome : Kribbelgefühl auf der Kopfhaut, Kopfschmerzen (64,7 %), Tinnitus (24,7 %), Müdigkeit (21,1 %), grippeähnliche Symptome2)
Akute Phase (Phase des Auftretens der Augensymptome)
Akuter Sehverlust : Bilaterale seröse Netzhautablösung (57,6 % der akuten VKH)2)
Leichte Vorderkammerentzündung und Glaskörpertrübung, Rötung und Schwellung der Papille (ca. 70 %)
Aderhautfalten : In schweren Fällen Aderhautablösung und flache Vorderkammer
Chronische Phase (Rezidivphase)
Granulomatöse Entzündung des vorderen Augenabschnitts steht im Vordergrund
Speckige KP, Koeppe-/Busacca-Knötchen, hintere Synechien der Iris
Bei 33,3 % der chronischen VKH tritt ein Sunset-Glow-Fundus auf2)
Spätphase (Erholungsphase)
Sunset-Glow-Fundus (sunset glow fundus): Verlust des Aderhautmelanins
Sugiura-Zeichen: Depigmentierung des Hornhautlimbus (etwa 1 Monat nach Beginn)
Hautsymptome (ca. 20 %): Vitiligo, Alopezie, Poliosis (nach einigen Monaten)
Makuläre Pigmentansammlung, verstreute chorioretinale Depigmentierungsherde
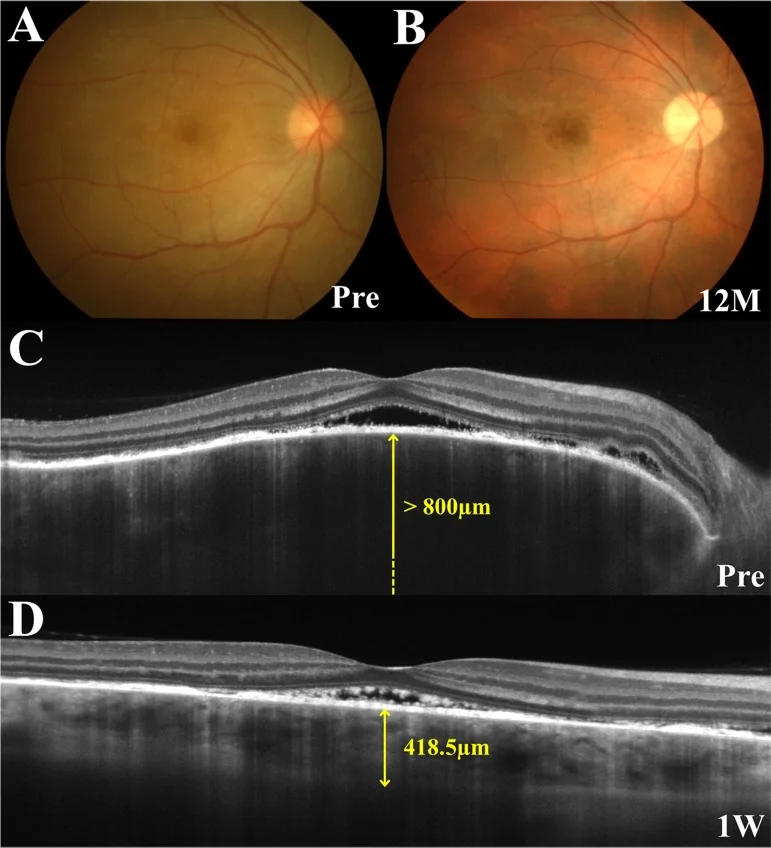
In der Prodromalphase treten Kribbeln der Kopfhaut, Kopfschmerzen und Tinnitus auf, gefolgt von einer plötzlichen Sehverschlechterung und verschwommenem Sehen 1–2 Wochen später, gleichzeitig oder nacheinander auf beiden Augen. Im OCT sind septierte Strukturen innerhalb der akuten Netzhautablösung charakteristisch.
Meningeale Reizsymptome (Kopfschmerzen, Nackensteifigkeit) treten bei den meisten Patienten früh im Krankheitsverlauf auf. Schallempfindungsschwerhörigkeit (wenig bemerkt, aber in Tests nachweisbar). Hautsymptome (Vitiligo, Alopezie, Poliosis) treten einige Monate nach Beginn auf und werden bei etwa 20 % der Fälle beobachtet.
Bei einem Rückfall ist die seröse Netzhautablösung des hinteren Segments, wie beim ersten Schub, selten; die granulomatöse Entzündung des vorderen Segments steht im Vordergrund. Es treten speckige Hornhautendothelbeschläge (KP), Koeppe- und Busacca-Knötchen auf, und hintere Synechien der Iris sind häufig. Prodromalsymptome wie Augenrötung und Verschwommensehen können auftreten.
Die CD4-positive T-Zell-Autoimmunreaktion gegen Melaninproteine (Tyrosinase-Familie: Tyrosinase, TRP-1, TRP-2, gp100) steht im Zentrum der Pathologie 3). Zielorgane sind Melanozyten der Uvea (Aderhaut), des Zentralnervensystems (Meningen), des Innenohrs und der Haut; die granulomatöse Entzündung der Aderhaut ist die primäre Frühveränderung.
Als genetische Prädisposition ist eine starke Assoziation mit HLA-DR4 (insbesondere DRB10405) bekannt 3). Auch ein Kopplungsungleichgewicht mit HLA-DPB10501 wurde berichtet 3). Es besteht die Hypothese, dass Virusinfektionen (EBV, CMV) durch molekulare Mimikry eine Autoimmunität auslösen.
Hauptrisikofaktoren:
| Typ | Kriterien |
|---|---|
| Komplette VKH-Erkrankung | Augenbefunde + neurologische/auditive Befunde + Hautmanifestationen, alle vorhanden |
| Inkomplette VKH-Erkrankung | Augenbefunde vorhanden, aber Fehlen von neurologischen/auditiven Befunden oder Hautmanifestationen |
| Wahrscheinliche VKH-Erkrankung | Nur Augenbefunde (isolierte Uveitis) |
Im Frühstadium sind keine Hautveränderungen zu sehen, daher sind die meisten Fälle unvollständig. Später, wenn Hautveränderungen auftreten, werden sie zu vollständigen Fällen. Die bilaterale seröse Netzhautablösung im Frühstadium ist charakteristisch, und die Diagnose ist einfach, wenn Prodromalsymptome wie Kopfschmerzen oder extraokuläre Symptome wie Tinnitus vorliegen 1). Bei atypischen Fällen (Papillenödem-Typ, einseitig) ist die Liquoruntersuchung entscheidend für die Diagnose.
Liquoruntersuchung: Lymphozytäre Pleozytose. Hält bis zu 8 Wochen an. Am nützlichsten zur Diagnosebestätigung.
Fluoreszenzangiographie (FA): Früh: fleckige Hypofluoreszenz durch verzögerte Aderhautfüllung → mittlere Phase: punktförmige Leckage (pinpoint leakage) → späte Phase: Farbstoffansammlung im SRD-Bereich. Hyperfluoreszenz der Papille (ca. 70 %) 1).
ICG-Angiographie: Früh: fleckige verzögerte Aderhautfüllung und Farbstoffaustritt aus Aderhautgefäßen, mittlere bis späte Phase: verstreute hypofluoreszente Flecken. Auch die Unschärfe mittlerer und großer Gefäße aufgrund einer Aderhautdurchblutungsstörung ist charakteristisch.
OCT: Erkennung und Überwachung der serösen Netzhautablösung. Früh: deutliche Aderhautverdickung. Mit EDI-OCT kann der Aderhautquerschnitt detailliert beobachtet werden. Im SRD können fibrinmembranartige Strukturen und Septen vorhanden sein 1).
B-Bild-Sonographie: Aderhautverdickung. Nützlich zur Abgrenzung von der hinteren Skleritis 1).
HLA-Klasse II: HLA-DR4 (Hilfsuntersuchung). Positivitätsrate 80 %, aber geringe Spezifität (auch bei Gesunden 25 % positiv).
ERG (Elektroretinogramm): Im chronischen Stadium Abnahme der photopischen und skotopischen Amplituden 3).
| Differenzialerkrankung | Abgrenzungsmerkmale |
|---|---|
| Hintere Skleritis | Einseitig, T-Zeichen im Ultraschall, Schmerz bei Augenbewegung |
| Multifokale posteriore Pigmentepitheliopathie (MPPE) | Einseitig, bewegliche bullöse seröse Netzhautablösung |
| Akute posteriore multifokale plakoide Pigmentepitheliopathie (APMPPE) | FA-Inversionsphänomen (frühe Hyperfluoreszenz → späte Hypofluoreszenz), junge Patienten, vorausgegangene Infektion |
| Zentrale seröse Chorioretinopathie (CSCR) | Einseitig, männlich bevorzugt, Steroide sind ein verschlimmernder Faktor3) |
| Sympathische Ophthalmie | Abgrenzung durch Vorgeschichte einer perforierenden Augenverletzung oder intraokularen Operation. Pathologisch als dieselbe Erkrankung angesehen1) |
| Idiopathisches uveales Exsudationssyndrom | Assoziation mit Nanophthalmus, Achtung auf Kammerwinkelblock |
Nicht obligatorisch, aber bei schwierig zu diagnostizierenden atypischen Fällen (Papillenödem-Typ, einseitiger Fall) entscheidend. Die lymphozytenbetonte Pleozytose hält bis zu 8 Wochen an, daher erhöht die Durchführung zum geeigneten Zeitpunkt nach Beginn die diagnostische Genauigkeit. Bei typischen Fällen mit eindeutiger bilateraler seröser Netzhautablösung kann die Diagnose oft allein anhand der klinischen Befunde gestellt werden.
Die Steroid-Pulstherapie gefolgt von einer oralen hochdosierten Ausschleichung ist der Standard. Ohne Behandlung kommt es in vielen Fällen zu Rezidiven oder Chronifizierung mit schwerer Sehbehinderung. Da es in der heutigen Medizin keinen Indikator gibt, um spontan heilende Fälle von rezidivierenden/chronischen Fällen zu unterscheiden, wird nach Möglichkeit eine hochdosierte Steroidtherapie empfohlen.
Leitlinienrepräsentatives Fallrezeptbeispiel1):
Oraler Reduktionsplan (Prednison 5 mg Tabletten):
| Dosis | Dauer |
|---|---|
| 200 mg/Tag | 2 Tage |
| 150 mg/Tag | 2 Tage |
| 100 mg/Tag | 2 Tage |
| 80 mg/Tag | 2 Tage |
| 60 mg/Tag | 4 Tage |
| 40 mg/Tag | 10 Tage |
| 30 mg/Tag | 2 Wochen |
| 20 mg/Tag | 4 Wochen |
| 15 mg/Tag | 4 Wochen |
| 10 mg/Tag | 4 Wochen |
| 5 mg/Tag | 4 Wochen |
| 5 mg/Tag (jeden zweiten Tag) | 4 Wochen |
Die Dosisreduktion sollte langsam erfolgen, und die Behandlung sollte auch ohne Rückfall über mindestens 6 Monate ausgeschlichen werden. Bei einer Behandlung von weniger als 6 Monaten beträgt die Rückfallrate 58,8 %, bei 6 Monaten oder mehr sinkt sie auf 11,1 % 5). Bei einem Rückfall die Steroiddosis erhöhen und über einen längeren Zeitraum als zuvor ausschleichen. Bei einer Steroid-Monotherapie kommt es bei 44 % zu einem Rückfall und bei 59 % zu einem Sunset-Glow-Fundus 2).
Behandlungsdosis für pädiatrische Patienten: Bei Kindern mit ausreichendem Alter und Gewicht wird entweder eine intravenöse Infusion von Methylprednisolon 500 mg/Tag über 3 Tage (Pulstherapie) oder eine orale Ausschleichung von Prednisolon beginnend mit 0,5–1,0 mg/kg/Tag durchgeführt. Die Dosis wird unter Beachtung der Nebenwirkungen reduziert.
Ciclosporin (Neoral®): 3 mg/kg/Tag (bei 60 kg Körpergewicht 180 mg/Tag in 2 Dosen). Regelmäßige Bestimmung der Talkonzentration erforderlich. Ermöglicht eine Reduktion der Steroiddosis. Anwendung bei verlängertem Verlauf 1). Achtung auf Nebenwirkungen wie Infektanfälligkeit, Nieren- und Leberfunktionsstörungen.
Methotrexat (MTX): 25 mg/Woche oral. Beginn mit 15 mg/Woche für 2 Wochen, dann Dosissteigerung 2).
Mycophenolatmofetil (MMF): 1,5 g zweimal täglich oral. Beginn mit 500 mg zweimal täglich und schrittweise Steigerung 2).
Die FAST-Studie (NCT01829295) ist eine RCT bei nicht-infektiöser Uveitis (Subanalyse von 93 VKH-Fällen von 216 Patienten). 49 Patienten wurden randomisiert MTX und 44 MMF zugeteilt, mit Prednison 1 mg/kg/Tag (max. 60 mg/Tag) zu Beginn und anschließender Dosisreduktion (Ziel ≤ 7,5 mg/Tag nach 6 Monaten).
6-Monats-Primärendpunkt (VKH-Subanalyse) :
| Indikator | MTX-Gruppe | MMF-Gruppe | P-Wert |
|---|---|---|---|
| Behandlungserfolgsrate | 80,4 % (37/46 Fälle) | 64,1 % (25/39 Fälle) | 0,10 |
| Abnahme der fovealen Netzhautdicke | −62,5 μm | −4,0 μm | 0,003 |
| SRD-Verschwindensrate | 86,3 % | 64,1 % | 0,02 |
| Sehverbesserung | Gleichwertig | Gleichwertig | 0,78 |
Die Gesamterfolgsrate der Behandlung betrug 74,7 % (62/85 Fälle). Bei akuter VKH war MTX hinsichtlich der Reduktion der zentralen Netzhautdicke (CST) und des Verschwindens der serösen Netzhautablösung (SRD) überlegen. Nach 12 Monaten behielten 91,3 % der MMF-Gruppe den Behandlungserfolg, und etwa die Hälfte (MTX 50,0 %, MMF 56,5 %) erreichte eine vollständige Prednison-Absetzung 2). Nach initialem Therapieversagen führte ein Wechsel von MMF zu MTX in 81,8 % der Fälle zum Erfolg 2). Bei initialer MMF-Kombinationstherapie behielten 93 % der akuten VKH-Patienten einen Visus von 20/20, und es wurden keine Rezidive oder Bildung eines Sunset-Glow-Fundus berichtet 2).
Bei Patienten, bei denen eine systemische Steroidgabe schwierig ist (Ältere, Schwangere, Diabetiker, psychiatrische Vorgeschichte), sollte eine subtenonale hintere Injektion von Triamcinolonacetonid in Betracht gezogen werden 1).
Begleitkatarakt: Durch hochdosierte Steroidanwendung tritt häufig ein Steroidkatarakt auf. Bei vollständiger Remission ist das Risiko ähnlich wie bei einer Standardoperation. Die IOL-Implantation ist unproblematisch. Falls später eine filtrierende Operation erforderlich sein könnte, sollte ein kornealer Zugang unter Schonung der oberen Bindehaut gewählt werden.
Sekundärglaukom / Steroidglaukom: Drucksenkende Augentropfen (Betablocker, PG-Analoga, Carboanhydrasehemmer) → orale CAI → D-Mannitol-Infusion in dieser Reihenfolge anwenden. Trabekulotomie (wirksam bei Steroidglaukom), falls unzureichend, Trabekulektomie durchführen.
Auch ohne Rezidiv wird empfohlen, die Dosis über mindestens 6 Monate langsam zu reduzieren. Ein Absetzen in weniger als 6 Monaten führt zu einer hohen Rezidivrate von etwa 58,8 %, während eine Fortsetzung über 6 Monate die Rate auf 11,1 % senkt 5). Bei einem Rezidiv die Dosis langsamer reduzieren als beim vorherigen Mal und die Hinzunahme eines Immunsuppressivums erwägen.
Ciclosporin (Neoral® 3 mg/kg/Tag) wird als steroidsparendes Mittel bei persistierenden Fällen eingesetzt 1). Die FAST-Studie bestätigte die Wirksamkeit von Methotrexat (MTX 25 mg/Woche) und Mycophenolatmofetil (MMF 1,5 g × 2-mal/Tag). Bei akuter VKH zeigte MTX einen überlegenen Trend bei der Reduktion der fovealen Netzhautdicke und der SRD-Verschwinderate 2).
Die CD4-positive T-Zell-vermittelte Autoimmunreaktion gegen Melaninproteine (Tyrosinase, TRP-1, TRP-2, gp100) ist das zentrale pathologische Geschehen 3). Zielzellen sind Melanozyten der Uvea (Aderhaut), des Zentralnervensystems (Meningen), des Innenohrs und der Haut; die granulomatöse Entzündung der Aderhaut ist die primäre Frühveränderung.
Als genetische Prädisposition ist eine starke Assoziation mit HLA-DR4 (insbesondere DRB10405) bekannt 3), und auch ein Kopplungsungleichgewicht mit HLA-DPB10501 wurde berichtet 3). Etwa die Hälfte der Patienten zeigt innerhalb von zwei Wochen vor Ausbruch grippeähnliche Symptome, was die Hypothese stützt, dass eine Virusinfektion (EBV, CMV) durch molekulares Mimikry eine Autoimmunität auslöst.
Interpretation bildgebender Befunde: In der ICG zeigen sich aufgrund von Aderhautdurchblutungsstörungen eine Unschärfe mittlerer und großer Gefäße sowie hypofluoreszente Flecken. Die frühe deutliche Aderhautverdickung im EDI-OCT spiegelt ein Aderhautstromaödem wider. Der Melaninverlust in der chronischen Phase führt zum Sonnenuntergangsfundus 3).
Etwa 25 % der Fälle zeigen trotz Steroid-Pulstherapie eine persistierende Entzündung; das Fortbestehen der Aderhautentzündung führt zu einer allmählichen Ausbreitung der Netzhaut-Aderhaut-Atrophie und bei einigen Patienten zu schwerer Sehbehinderung. Subjektive Symptome wie leichte Metamorphopsie oder Farbsehstörungen bleiben oft bestehen.
Klinische Bedeutung der FAST-Studie 2): Erste direkte Vergleichs-RCT von MTX vs. MMF bei nichtinfektiöser Uveitis. Die 93 VKH-Fälle waren die größte Untergruppe der Studie und zeigten die Wirksamkeit einer frühen Einführung von Immunsuppressiva bei akuter VKH. Weitere Nachbeobachtungsstudien werden erwartet.
Bedeutung der frühen Einführung von Immunsuppressiva 2): Bei alleiniger Steroidtherapie erleiden 44 % ein Rezidiv und 59 % entwickeln einen Sonnenuntergangsfundus. Die frühe Kombination von Steroiden und Antimetaboliten kann die Rezidivrate und die Inzidenz des Sonnenuntergangsfundus senken. Das Konzept des „therapeutischen Fensters“ – der frühzeitige Beginn von Steroiden zu Beginn der Erkrankung verhindert das Fortschreiten in die chronisch-rezidivierende Phase und reduziert die Notwendigkeit einer langfristigen Immunsuppression – gewinnt an Bedeutung.
Fortschritte in der Bildgebung3): Die quantitative Beurteilung der Aderhautdicke mittels EDI-OCT und der Nachweis einer okkulten Aderhautentzündung mittels ICG werden für die Therapieüberwachung genutzt. Die quantitative Messung hyperreflektiver Aderhautherde (HCF) mittels En-face-OCT könnte ein Biomarker für die Aktivität der VKH-Krankheit sein.
Trends bei den überarbeiteten Diagnosekriterien2): Eine frühe/späte Klassifikation unter Einbeziehung von OCT, FA und ICG wurde vorgeschlagen, was die diagnostische Genauigkeit verbessern dürfte. Die derzeitigen überarbeiteten Kriterien von 2001 weisen Einschränkungen bei der Erkennung einer frühen VKH auf.