Sarkoidose ist eine systemische entzündliche Erkrankung unbekannter Ursache, die zur Bildung nicht-verkäsender epitheloidzelliger Granulome in verschiedenen Organen führt. Histopathologisch sind Granulome ohne Nekrose, bestehend aus Epitheloidzellen und Riesenzellen, charakteristisch. Häufige Lokalisationen sind Lunge, mediastinale Lymphknoten, Augen und Haut, aber auch Herz, Gehirn, Knochen, Nieren, Magen-Darm-Trakt usw. können betroffen sein.
1878 berichtete Sir Jonathan Hutchinson erstmals über die Erkrankung als Hauterkrankung. 1909 beschrieb der dänische Augenarzt Heerfordt Fälle mit Uveitis, Parotitis und Fieber, die als Heerfordt-Syndrom bezeichnet werden 9).
Es ist die häufigste Ursache für Uveitis. In einer epidemiologischen Studie von 2002 machte sie 13,3 % aller Uveitiden aus, 2009 waren es 10,7 % (jeweils die häufigste Ursache) 1). 20–50 % der Sarkoidose-Patienten entwickeln eine Augenbeteiligung 2), und bei 30–40 % sind Augensymptome die Erstmanifestation. Über 85 % der Fälle sind bilateral 2).
Das Erkrankungsalter zeigt bei Männern einen Gipfel in den 20ern, bei Frauen eine bimodale Verteilung mit Gipfeln in den 20ern und den 50–60ern. Frauen sind häufiger betroffen (Männer:Frauen-Verhältnis 1:1,8) 1), und nach dem 50. Lebensjahr steigt der Frauenanteil.
Die pädiatrische Sarkoidose unterscheidet sich von der Erwachsenenform 3). Die frühkindliche Form (≤5 Jahre) entspricht dem Blau-Syndrom mit NOD2-Genmutation (R334W, R334Q) und ist durch die Trias Arthritis, Dermatitis und Uveitis gekennzeichnet. Die im Alter von 8–15 Jahren auftretende Erwachsenenform wird auf eine überschießende Immunantwort auf Umweltantigene zurückgeführt 3).
QWarum ist die sarkoide Uveitis in Japan häufig?
A
Sie ist die häufigste Ursache für Uveitis, während ihr Anteil in Europa, den USA und Südostasien geringer ist. Es wird vermutet, dass genetische Faktoren (HLA-DRB1-Allele) und Umweltfaktoren eine Rolle spielen, die genaue Ursache ist jedoch unbekannt 1).
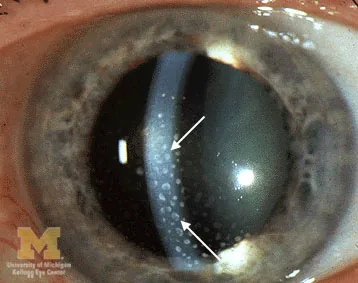
Trobe JT. Keratic precipitates. The Eyes Have It — Anterior Uveitis. University of Michigan Kellogg Eye Center. Via Wikimedia Commons (File:Keratic-precipitates.jpg). License: CC BY 3.0.
Große, gelblich-weiße, fettige granulomatöse Ablagerungen auf dem Hornhautendothel, mit weißen Pfeilen markiert. Entspricht den im Abschnitt „2. Hauptsymptome und klinische Befunde“ beschriebenen Mutton-Fat-KPs.
Die Augensymptome variieren je nach Lokalisation und Schweregrad der Entzündung. Verschwommenes Sehen ist am häufigsten, gefolgt von Floatern (durch Glaskörpertrübung), verminderter Sehschärfe, Photophobie, Rötung und Augenschmerzen. Bei chronischen Formen kann der Verlauf asymptomatisch sein, was die Diagnose verzögert 2). Die Augensymptome können den Allgemeinsymptomen um mehrere Jahre vorausgehen.
:::caution Vorsicht auch bei fehlenden Symptomen
Bei chronischen Verläufen sind die Symptome gering. Erst durch regelmäßige augenärztliche Untersuchungen kann eine aktive Entzündung festgestellt werden.
:::
Eine Schwellung der Tränendrüse durch ein Granulom kann zu einer Keratokonjunktivitis sicca führen. Es werden auch Bindehautknötchen (oft asymptomatisch), Skleritis (selten, nicht-nekrotisierend) und Granulome der Lidhaut beobachtet. Es kann auch zu Hirnnervenlähmungen wie einer Fazialisparese kommen.
Die Ätiologie ist unbekannt. Es wird angenommen, dass bei genetisch prädisponierten Personen eine überschießende Immunantwort ausgelöst wird, wenn sie Umweltantigenen (Inhalationsantigene, Infektionserreger) ausgesetzt sind.
Eine Beteiligung von Cutibacterium acnes (früher Propionibacterium acnes) wurde berichtet. Auch eine Beteiligung von Tuberkulose-Bakterien-DNA und verschiedener Viren wurde vermutet 2). Als genetische Prädisposition ist eine Assoziation mit HLA-DRB1 bekannt 1), und das Risiko ist bei familiären Fällen erhöht 2).
Risikofaktoren
Beschreibung
Ethnie
Häufiger bei Afroamerikanern (etwa 10-mal häufiger als bei Weißen) und auch häufig bei Nordeuropäern
Geschlecht
Etwas häufiger bei Frauen (Verhältnis Männer:Frauen 1:1,8)
Alter
Männer in den 20ern, Frauen in den 20ern und in den 50ern-60ern
HLA
Assoziation mit HLA-DRB1-Allelen1)
Familienanamnese
Erhöhtes Risiko bei Vorliegen einer Sarkoidose bei einem Verwandten ersten Grades2)
Bei der frühkindlichen Form (EOS) führen NOD2-Mutationen (R334W, R334Q) zu einer Überaktivierung des NF-κB-Signalwegs und zum Auftreten pathologischer Th17-Zellen3).
:::tip Prävention und Alltag
Derzeit gibt es keine etablierte Präventionsmethode. Für eine Früherkennung ist es wichtig, bei anhaltendem verschwommenem Sehen oder Flugkörpern einen Augenarzt aufzusuchen.
:::
Das Prinzip der Diagnose ist die Gesamtbeurteilung von klinischen Befunden, Untersuchungsergebnissen und histologischen Befunden.
:::caution Vorsicht bei systemischer Steroidgabe vor der Diagnose
Eine systemische Steroidgabe kann Läsionen verkleinern und Gewebebiopsieergebnisse falsch-negativ machen. Vermeiden Sie systemische Steroide bis zur Diagnosesicherung, es sei denn, es liegt ein Notfall vor.
:::
QKann eine Sarkoidose ausgeschlossen werden, wenn der ACE-Wert im Blut normal ist?
A
Nein. Die Sensitivität beträgt 73%, sodass etwa ein Viertel der Fälle normale Werte aufweist. Unter Steroidtherapie oder Einnahme von ACE-Hemmern kann es zu falsch negativen Ergebnissen kommen. Eine Gesamtbewertung von klinischen Befunden, Bildgebung und anderen Tests ist erforderlich 2).
QWie unterscheidet man Sarkoidose von malignem Lymphom?
A
Beide können Aderhautläsionen und Lymphknotenschwellungen verursachen. Der IL-10/IL-6-Quotient im Kammerwasser oder Glaskörper (>1 deutet auf Lymphom hin) und das Anreicherungsmuster im FDG-PET helfen bei der Unterscheidung. Zur definitiven Diagnose ist eine Gewebebiopsie erforderlich 5).
Indikationen für systemische Gabe 1): schwere, auf lokale Therapie resistente Vorderkammerentzündung, starke Glaskörpertrübung, ausgedehnte Chorioretinitis, Netzhautvaskulitis, Makulaödem, Papillenödem, Granulom.
Prednisolon 0,5–1,0 mg/kg/Tag für 2–4 Wochen beginnen, dann alle 4–8 Wochen um 5–10 mg/Tag reduzieren 1). Die Gesamtbehandlungsdauer kann 6 Monate bis über 1 Jahr betragen 1).
Beispiel für einen Reduktionsplan:
Dosis
Dauer
30 mg/Tag
2 Wochen
20 mg/Tag
1 Monat
15 mg/Tag
1 Monat
10 mg/Tag
1 Monat
7,5 mg/Tag
1 Monat
5 mg/Tag
1 Monat
5 mg/Tag (jeden zweiten Tag)
1 Monat
:::caution Hinweise zur Langzeitanwendung von Steroiden
Achten Sie auf das Risiko von Osteoporose, Diabetes, Bluthochdruck und Infektionen. Bei Langzeitanwendung sollte die gleichzeitige Gabe von Magenschutzmitteln und Bisphosphonaten in Betracht gezogen werden.
:::
Adalimumab (Humira®) : 40 mg alle 2 Wochen subkutan2, 4). Anti-TNF-α-Antikörper, zugelassen für nichtinfektiöse Uveitis. Die VISUAL-I- und VISUAL-II-Studien bestätigten die Wirksamkeit zur Rezidivprophylaxe4).
Infliximab (Remicade®) : 5 mg/kg i.v. alle 8 Wochen1). Zugelassen für refraktäre retinale Uveitis bei Morbus Behçet; Off-Label-Anwendung bei Sarkoidose.
Etanercept hat eine geringe Wirksamkeit1). Vor Biologika-Gabe ist ein Screening auf Tuberkulose und Hepatitis B obligatorisch4).
Kataraktoperation : Durchführung in der entzündungsfreien Phase. Bei unvollständiger Entzündungsfreiheit Operation in relativ ruhiger Phase unter oraler Steroidbegleitung. Falls später eine filtrierende Operation in Frage kommt, obere Bindehaut schonen und Hornhautschnitt wählen.
Sekundärglaukom : Behandlung mit drucksenkenden Augentropfen (PG-Analoga, Betablocker, Carboanhydrasehemmer, Alpha-2-Agonisten) → orale CAI → D-Mannitol-Infusion. Die Trabekulotomie ist besonders bei Steroidglaukom wirksam. Bei unzureichender Wirkung Trabekulektomie durchführen.
Netzhautphotokoagulation : Durchführung bei avaskulären Arealen infolge obliterierender Vaskulitis. Direkte Koagulation retinaler Mikroaneurysmen.
Bei leichten Entzündungen des vorderen Augenabschnitts ist dies möglich. Bei Läsionen des hinteren Augenabschnitts (zystoides Makulaödem, ausgedehnte Retinochoroiditis, Optikusneuropathie) ist eine subtenonale Injektion oder systemische Behandlung erforderlich.
QIst die Sarkoid-Uveitis heilbar?
A
Etwa 2/3 der Fälle haben einen gutartigen, selbstlimitierenden Verlauf mit guter Sehprognose ohne systemische Steroide. Einige Fälle werden chronisch und erfordern eine Langzeittherapie. Entzündungen des vorderen Augenabschnitts haben eine gute Prognose, während wiederholte Befall des hinteren Augenabschnitts zu Netzhautdegeneration und Optikusatrophie führen kann, was zu schweren Sehstörungen führt.
Die Granulombildung erfolgt durch folgende Immunkaskade 2):
Antigenerkennung über TLR2 → Makrophagenaktivierung
Produktion von IL-6, IL-12, IL-18, TNF-α
Th1-Differenzierung von CD4+ T-Zellen → Produktion von IFN-γ und IL-2. Th17 → IL-17
Funktionsstörung regulatorischer T-Zellen → Verstärkung der Th1-Antwort → Granulombildung
Histologische Merkmale von Granulomen: Ansammlungen von nicht-verkäsenden Epitheloidzellen und Lymphozyten. Asteroidkörperchen und Schaumann-Körperchen in mehrkernigen Riesenzellen. Ringförmige Fibrose in der Umgebung 2).
Wichtigste intraokulare Pathologien: Granulomablagerung im Trabekelwerk → Behinderung des Kammerwasserabflusses → sekundäres Glaukom 6). Granulomatöse Infiltration der Netzhautgefäßwände → Periphlebitis → Bildung von wachsartigen Exsudaten. Erhöhte VEGF-Produktion im entzündlichen Milieu → choroidale Neovaskularisation10).
Übermäßige Vitamin-D-Produktion (erhöhte Synthese von 1,25(OH)₂D₃ durch Makrophagen) kann ebenfalls zu Hyperkalziurie und Hyperkalzämie führen 2).
Bei der frühkindlichen Form (NOD2-Mutation) fördern eine Überaktivierung des NF-κB-Signalwegs und das Auftreten pathologischer Th17-Zellen die Granulombildung 3).
:::danger Informationen zum Forschungsstadium
Einige der in diesem Abschnitt beschriebenen Behandlungen und Forschungsergebnisse befinden sich noch im Forschungsstadium. In der klinischen Praxis sollte die Entscheidung in Absprache mit einem Facharzt getroffen werden.
:::
Anwendung von Faricimab bei refraktärem zystoidem Makulaödem12): Dualer Inhibitor von VEGF und Ang-2. Im Bericht von Lin 2025 zeigte eine 82-jährige Frau mit steroidresistentem CME nach zwei Injektionen eine Verbesserung des BCVA in beiden Augen (rechts 20/200→20/50, links 20/400→20/63) und eine deutliche Verbesserung der fovealen Netzhautdicke (rechts 562→371 μm, links 717→286 μm). Dies ist der weltweit erste Bericht über die Anwendung bei okulärer Sarkoidose12).
Zusammenhang mit Immun-Checkpoint-Inhibitoren11): Read 2025 berichtete über einen Patienten mit schwerer okulärer Sarkoidose in der Vorgeschichte, der über 2 Jahre Pembrolizumab erhielt, ohne dass es zu einem Augenrezidiv kam. Dies ist ein Fallbericht, der die Möglichkeit einer sicheren Anwendung unter sorgfältiger Überwachung aufzeigt.
Zusammenhang zwischen Sarkoidose und malignen Tumoren (Sarkoid-Lymphom-Syndrom)5): Eine Störung der immunregulatorischen Mechanismen kann für die Entstehung eines Lymphoms prädisponieren, daher ist bei der Langzeitnachsorge auf das Auftreten von Tumoren zu achten.
Fortschritte in der multimodalen Bildgebung2): EDI-OCT stellt Aderhautgranulome als homogene, hyporeflektive, gut abgegrenzte Läsionen dar. OCTA (optische Kohärenztomographie-Angiographie) ermöglicht den Nachweis von choroidalen Kapillarblutflussdefekten. ICGA (Indocyaningrün-Angiographie) ist nützlich für den Nachweis potenzieller Aderhautgranulome und die Beurteilung des Therapieansprechens.
Langzeitprognose der pädiatrischen Sarkoidose3): In einer Serie von 52 pädiatrischen Fällen des Erwachsenentyps (mediane Nachbeobachtung 11,5 Jahre) behielten 50 % die aktive Erkrankung bis ins Erwachsenenalter. 19 % der Patienten mit Remission im Kindesalter erlitten im Erwachsenenalter ein Rezidiv. Eine lebenslange Nachbeobachtung wird empfohlen.
Neuer Bericht über entzündliche choroidale Neovaskularisation10): Bei einem 14-jährigen Kind wurde trotz gleichzeitiger Anwendung von Adalimumab eine Progression einer peripapillären CNV berichtet, die eine zusätzliche intravitreale Anti-VEGF-Injektion erforderte.
Berkowitz ST, Brock AL, Reichstein DA. Chorioretinal biopsy-proven ocular sarcoidosis in a patient with a history of B-cell lymphoma. Case Rep Ophthalmol. 2021;12(2):438-445. doi:10.1159/000512694. PMID:34054498; PMCID:PMC8136327.
Sarcoidosis and inflammatory glaucoma. Oman J Ophthalmol. 2011;4(1):5.
Oyeniran E, Katz D, Kodati S.. Isolated Optic Disc Granuloma as a Presenting Sign of Sarcoidosis. Ocul Immunol Inflamm. 2024;32(2):175-177. doi:10.1080/09273948.2022.2127783. PMID:36223603; PMCID:PMC10090223.
Riccardi M, Contento R, Christensen C, Brady A, Swan RL, Swan RT. A Case of Sarcoid Uveitis Diagnosed With Mammography Two Months After Normal Chest Imaging. Case reports in ophthalmological medicine. 2025;2025:8871004. doi:10.1155/crop/8871004. PMID:39975857; PMCID:PMC11839256.
Nakamura M, Suzuki K, Yoshida E, Watanabe K, Sasage H, Dohi K, Suzuki S.. A Case of Incomplete Heerfordt Syndrome Diagnosed Following Fever Onset. Cureus. 2025;17(10):e94234. doi:10.7759/cureus.94234. PMID:41210040; PMCID:PMC12596090.
Zong Y, Wang K, Zhang T, Xu G.. Pediatric peripapillary choroidal neovascularization secondary to ocular sarcoidosis: a long-term follow-up case. BMC Ophthalmol. 2025;25(1):422. doi:10.1186/s12886-025-04266-7. PMID:40702433; PMCID:PMC12285086.
Read C, Bhatia S, Totonchy M.. Programmed cell death 1 blockade in the setting of severe ocular sarcoidosis: Cancer immunotherapy in a patient with autoimmunity. JAAD Case Rep. 2025;62:43-45. doi:10.1016/j.jdcr.2025.04.017. PMID:40661112; PMCID:PMC12256288.
Lin TH, Lin HY, Chuang YH, Tseng PC.. Faricimab as Treatment for Sarcoid Uveitis With Refractory Cystoid Macular Edema. J Vitreoretin Dis. 2025:24741264251358628. doi:10.1177/24741264251358628. PMID:40765984; PMCID:PMC12318965.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.