Einfache Episkleritis
Häufigkeit: Häufiger
Beginn: Plötzlich
Verlauf: Erreicht nach etwa 12 Stunden den Höhepunkt und klingt nach 2–3 Tagen ab
Befund: Fächerförmige (ca. 67%) oder diffuse (ca. 33%) Rötung

Episkleritis ist eine benigne, selbstlimitierende Rötung des episkleralen Gewebes. Es handelt sich um eine Entzündung der oberflächlichen Gefäße, wie des Tenon-Kapsel-Gefäßplexus. Im Vergleich zur Skleritis, die tiefere Gefäße betrifft, sind die Schmerzen geringer und die Sehkraft kaum beeinträchtigt. Meist ist sie idiopathisch und rezidivierend, mit Tendenz zu beidseitigem Auftreten. Die Inzidenz wird mit 41,0 pro 100.000 Personen pro Jahr und die Prävalenz mit 52,6 angegeben.
Sie gehört zu den relativ häufigen Ursachen für Rötung des Auges, wird aber oft mit Konjunktivitis oder Skleritis verwechselt und daher bei der Erstvorstellung nicht selten fehldiagnostiziert. Bei dieser Erkrankung ist das Sklerastroma selbst nicht betroffen, und es kommt fast nie zu schweren strukturellen Komplikationen wie einer Perforation. Bei rezidivierenden Fällen oder solchen mit systemischen Autoimmunerkrankungen wie rheumatoider Arthritis oder Granulomatose mit Polyangiitis sind jedoch die Behandlung der Grunderkrankung und eine langfristige Nachsorge erforderlich. Das Verständnis der Episkleritis nicht nur als isolierte Augenerkrankung, sondern als okuläre Manifestation einer systemischen Erkrankung ist entscheidend für das Rezidivmanagement und die Prognoseverbesserung.
Die Watson-Klassifikation wird häufig für die klinische Einteilung entzündlicher Erkrankungen der Sklera und Episklera verwendet. Sie unterscheidet drei Hauptgruppen nach Lokalisation: Episkleritis, anteriore Skleritis und posteriore Skleritis. Die anteriore Skleritis wird weiter nach Form in diffus, nodulär und nekrotisierend (entzündlich/nicht entzündlich) unterteilt. Ein wichtiger Unterschied zur anterioren Skleritis ist, dass es bei der Episkleritis keine nekrotisierende Form gibt und sie morphologisch nur in zwei Typen eingeteilt wird: einfach (diffuser Typ) und nodulär. Diese Klassifikation spiegelt die Entzündungstiefe (oberflächlich oder tief) sowie den Schweregrad von Verlauf und Prognose wider, sodass die Bestimmung des Krankheitstyps bei der Diagnose die Grundlage für die Behandlungsstrategie und Prognoseerklärung bildet. Die Episkleritis wird in dieser Klassifikation als die mildeste Gruppe mit der besten Prognose eingestuft.
Einfache Episkleritis
Häufigkeit: Häufiger
Beginn: Plötzlich
Verlauf: Erreicht nach etwa 12 Stunden den Höhepunkt und klingt nach 2–3 Tagen ab
Befund: Fächerförmige (ca. 67%) oder diffuse (ca. 33%) Rötung
Noduläre Episkleritis
Häufigkeit: Etwas seltener
Beginn: Langsam
Verlauf: Neigt zu länger anhaltenden Symptomen als die einfache Form
Befund: Umschriebener, beweglicher episkleraler Knoten in der Nähe des Hornhautlimbus
Die Sklera besteht aus drei Schichten: Episklera, Sklerastroma und Lamina fusca. Die Episklera ist ein gefäßführendes Bindegewebe über dem Sklerastroma und wird als fibroelastische Struktur zwischen Sklerastroma und Tenon-Kapsel verstanden. Sie besteht aus einer äußeren parietalen Schicht (oberflächliches episklerales Kapillarnetz) und einer inneren viszeralen Schicht (stark anastomosierendes Gefäßnetz), die beide aus den vorderen Ziliararterien stammen. Die meisten Nervenfasern stammen vom Nervus trigeminus. Die Episklera bildet zwischen den Ansätzen der geraden Augenmuskeln und dem Limbus einen episkleralen Gefäßplexus, der normalerweise von der Bindehaut verdeckt wird, sich aber bei Entzündung erweitert und eine lebhafte Rötung verursacht. Die Episklera wird nach hinten zum Auge hin allmählich dünner, und im hinteren Augenabschnitt dominiert die Tenon-Kapsel.
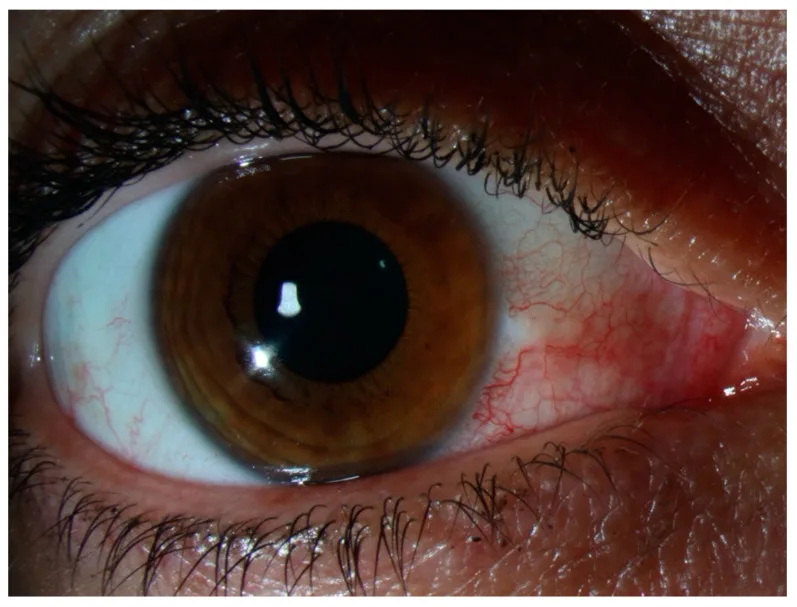
Es besteht kein Druckschmerz und kein Ausfluss. Bei starken Schmerzen oder deutlichem Ausfluss sollten Skleritis, infektiöse Konjunktivitis oder anteriore Uveitis erneut in Betracht gezogen werden. Die Symptome klingen meist innerhalb weniger Tage ab oder verschwinden vollständig, ohne die Sehfunktion zu beeinträchtigen. Bei einem Rezidiv tritt es oft an derselben Stelle oder am kontralateralen Auge auf, und die Patienten bemerken es häufig als „das übliche rote Auge“. Starke Schmerzen, die den Nachtschlaf stören, oder starker Druckschmerz beim Berühren des Oberlids, wie bei einer Skleritis, treten bei einer Episkleritis in der Regel nicht auf.
Die Beobachtung von Lokalisation und Farbe der Rötung ist zentral für die Differenzialdiagnose. Die Rötung bei Episkleritis ist hellrot bis rosa, im Gegensatz zur dunkelroten (purpurroten) tiefen Rötung bei Skleritis.
| Befund | Episkleritis | Skleritis |
|---|---|---|
| Farbe der Rötung | Hellrot bis rosa | Dunkelrot (purpurrot) |
| Schmerz | Gering bis keiner | Stark ausstrahlend |
| Beweglichkeit des Knotens | Vorhanden | Nicht vorhanden |
Die Sehschärfe ist in der Regel normal. Bindehautödem, erhöhter Augeninnendruck, anteriore Uveitis und Keratitis sind seltene Begleiterscheinungen; treten sie auf, sollte an eine Skleritis oder andere Erkrankungen gedacht werden. Das Fehlen von Entzündungszeichen an der Lidbindehaut ist hilfreich zur Unterscheidung von einer Konjunktivitis. Während eine Skleritis durch Entzündungsausbreitung auf das umliegende Gewebe zu peripheren Hornhautinfiltraten, Ulzerationen oder anteriorer Uveitis führen kann, ist die Episkleritis selbstlimitierend und involviert nahezu nie benachbarte Gewebe. Im Spaltlampenmikroskop wird die Ebene des episkleralen Gefäßplexus identifiziert; selbst wenn eine rote erhabene Läsion vorliegt, aber die Skleragefäße nicht sichtbar sind, sollte auch die Möglichkeit einer neoplastischen Läsion in Betracht gezogen werden.
Die Episkleritis ist gekennzeichnet durch das Fehlen von Augenausfluss und die Lokalisation der Rötung in der Nähe des Hornhautlimbus. Die Konjunktivitis ist in der Regel schmerzlos, geht mit Augenausfluss einher, und die Rötung ist am stärksten im Fornix und nimmt zum Limbus hin ab. Im Spaltlampenmikroskop sind die episkleralen Gefäße unbeweglich, während die Bindehautgefäße beweglich sind – ein weiteres Unterscheidungsmerkmal. Details siehe Abschnitt „Diagnose und Untersuchungsmethoden“.
Die meisten Fälle sind idiopathisch (Ursache unbekannt); es wird berichtet, dass bei etwa 26–36 % aller Fälle eine systemische Erkrankung vorliegt. Auch bei idiopathischen Fällen wird eine Beteiligung immunologischer Mechanismen vermutet, die auf einer lymphozytenzentrierten, unspezifischen Entzündungsreaktion im oberflächlichen episkleralen Gefäßplexus beruht. Der rezidivierende Verlauf und die Tendenz zur bilateralen Beteiligung deuten auf eine zugrunde liegende systemische Immunregulationsstörung hin.
Kollagenosen/Autoimmunerkrankungen (am häufigsten rheumatoide Arthritis)1):
Vaskulitis:
Infektionen: Bakterien, Mykobakterien, Syphilis, Lyme-Borreliose, Herpesviren, Gürtelrose können Ursachen sein. Eine Episkleritis im Zusammenhang mit einem Herpes zoster ophthalmicus wird nicht als direkte Infektion, sondern als Immunantwort auf den Erreger angesehen. Es wurde auch ein Fall einer subkonjunktivalen Parasitose durch Dirofilaria repens berichtet, der fälschlicherweise als Episkleritis diagnostiziert wurde7).
Sonstiges: Gicht, Atopie, Fremdkörper, chemische Traumata, Medikamente (Topiramat, Pamidronat), sowie Berichte als Frühsymptom von COVID-19.
Ja, bei etwa einem Drittel der Patienten liegt eine begleitende systemische Erkrankung vor. Am häufigsten ist die rheumatoide Arthritis, aber auch die Granulomatose mit Polyangiitis (GPA) oder der Morbus Behçet können als Erstsymptom auftreten – Erkrankungen, bei denen eine frühzeitige Diagnose und Behandlung die Prognose beeinflusst. Bei wiederholten Rückfällen oder begleitenden systemischen Symptomen werden systemische Untersuchungen wie Rheumafaktor, antinukleäre Antikörper, ANCA und Urinuntersuchungen empfohlen.
Die Episkleritis wird hauptsächlich klinisch auf der Grundlage der Anamnese und der Spaltlampenuntersuchung diagnostiziert. Grundlegend ist die sorgfältige Beurteilung der Ebene der episkleralen Gefäße (oberflächlich oder tief), der Farbe der Hyperämie, des Vorhandenseins von Knötchen sowie von Ausdünnung oder Nekrose mittels Spaltlampe.
Phenylephrin 2,5 % verengt die Bindehautgefäße und ist nützlich zur Unterscheidung zwischen Konjunktivitis und Episkleritis. Phenylephrin 10 % verengt das oberflächliche episklerale Gefäßnetz, nicht jedoch das tiefe, sodass eine Unterscheidung zwischen Episkleritis und Skleritis möglich ist.
Der Reaktionstest mit 1:1000 verdünntem Epinephrin ist eine einfache Methode, um eine Beteiligung tiefer Gefäße zu beurteilen. Wenn die Hyperämie nach der Gabe zurückgeht, spricht dies für eine Episkleritis; bleibt sie bestehen, deutet dies auf eine Skleritis hin. Die Beurteilung erfolgt kombiniert anhand von drei Kriterien: Anzahl und Beweglichkeit der Knötchen, Vorhandensein von Schmerzen/Druckschmerz und Epinephrin-Reaktion.
Die Reaktionstests mit Epinephrin und Phenylephrin sind besonders hilfreich als ergänzende Diagnostik, wenn die Schichtstruktur der Hyperämie mit der Spaltlampe nicht direkt beurteilt werden kann oder bei kleinen Knötchen. Die Beobachtung 10–15 Minuten nach der Gabe zeigt, ob sich die oberflächlichen Gefäße verengen; bleibt eine Hyperämie der tiefen Gefäße bestehen, wird prioritär eine Skleritis behandelt.
Die Tenon-Kapsel-Entzündung wird ebenfalls als eine Form der Episkleritis angesehen, und die klinische Unterscheidung zwischen beiden ist schwierig. Die Beurteilung erfolgt anhand einer Kombination von Knotenverschieblichkeit, Vorhandensein von Schmerzen/Druckempfindlichkeit, Reaktion auf Epinephrin-Augentropfen und Fluorescein-Färbebefunden.
Bei einer einzelnen, milden Episkleritis ist eine umfassende systemische Abklärung nicht erforderlich. Bei wiederholten Rezidiven oder begleitenden systemischen Symptomen sollten die folgenden Untersuchungen in Betracht gezogen werden.
Bei Fällen, in denen eine Episkleritis als erstes Symptom einer Granulomatose mit Polyangiitis auftritt, kann eine Nierenfunktionsstörung gleichzeitig bestehen 3). Wenn sowohl eine Augenentzündung als auch eine Nierenfunktionsstörung vorliegen, sollte umgehend eine Untersuchung auf systemische Vaskulitis, einschließlich Granulomatose mit Polyangiitis, durchgeführt werden. Bei therapieresistenter oder rezidivierender Episkleritis ist es wünschenswert, in Zusammenarbeit mit der Rheumatologie und Inneren Medizin die Krankheitsaktivität zu bewerten und eine Behandlung der Grunderkrankung einzuleiten.
Zusätzlich zur Beurteilung mit der Spaltlampe können die optische Kohärenztomographie des vorderen Augenabschnitts (AS-OCT) zur Bewertung der Dicke der Episkleralschicht und des Gefäßverlaufs sowie die Ultraschalluntersuchung (B-Modus) zur Beurteilung der Skleradicke als ergänzende Diagnostik eingesetzt werden. Zum Ausschluss einer nekrotisierenden Skleritis oder zur Beurteilung einer hinteren Skleritis wird im Ultraschall das Vorhandensein eines T-Zeichens (Flüssigkeitsansammlung um die Sehnervenscheide) überprüft. Bei einer gewöhnlichen Episkleritis zeigen diese bildgebenden Verfahren oft keine spezifischen Befunde, sodass die Diagnose durch direkte Untersuchung mit der Spaltlampe in Kombination mit Anamnese und systemischer Abklärung gestellt wird.
Die Episkleritis heilt in den meisten Fällen ohne Behandlung innerhalb weniger Tage bis Wochen spontan aus. Die Aufklärung des Patienten über die Gutartigkeit und den natürlichen Verlauf der Erkrankung sowie die Notwendigkeit einer systemischen Abklärung und die Vermittlung von Beruhigung sind der erste Schritt der Behandlung. Kühlende Umschläge oder gekühlte künstliche Tränen sind wirksam zur Linderung von Symptomen wie Reizgefühl und Hitzegefühl. Bei leichten Fällen wird auf eine aktive medikamentöse Intervention verzichtet; durch kurzfristige Nachkontrollen im Abstand von wenigen Tagen wird die spontane Besserung bestätigt, wodurch Rebound-Effekte und Nebenwirkungen einer Behandlung vermieden werden können.
Niedrig dosierte Steroid-Augentropfen sind die erste Wahl. Häufig werden zusätzlich antibiotische Augentropfen zur Abgrenzung von einer Skleritis verabreicht.
Wenn die Reaktion auf die Tropfentherapie unzureichend ist, sollte ein Wechsel zur Untersuchung und Behandlung einer Skleritis in Betracht gezogen werden. Während Steroid-Augentropfen die Symptome schnell unterdrücken, wird darauf hingewiesen, dass eine langfristige oder wiederholte Anwendung das Rezidivrisiko erhöhen und einen „Rebound“-Reizzustand auslösen kann.
Die Behandlung wird in der Regel nach Abklingen der Symptome ausgeschlichen und abgesetzt; eine wahllose Fortführung ist zu vermeiden. Da die langfristige Anwendung von steroidhaltigen Augentropfen das Risiko eines steroidinduzierten Augeninnendruckanstiegs und einer subkapsulären Katarakt birgt, sollte nach 1–2 Wochen eine Besserung überprüft und die Dosis reduziert werden. Bei Rezidiven wird die Krankheitsaktivität jedes einzelnen Schubs individuell bewertet und die Optimierung der Behandlung der zugrunde liegenden systemischen Erkrankung priorisiert.
Bei einer mit Kollagenosen wie rheumatoider Arthritis assoziierten Episkleritis ist die Behandlung der Grunderkrankung entscheidend für die Prognose1). Bei Resistenz gegen die lokale Therapie wird orales Prednisolon (20–30 mg/Tag mit ausschleichender Dosierung) eingesetzt. Nur in seltenen Fällen ist eine systemische Steroidtherapie erforderlich, es sei denn, eine systemische Entzündungserkrankung liegt eindeutig vor.
Bei einer mit Granulomatose mit Polyangiitis assoziierten Episkleritis ist eine Remissionsinduktion mit Cyclophosphamid oder Rituximab wirksam3)4). Es gibt Berichte, dass Rituximab nach 6 Monaten eine höhere Remissionsrate aufweist als Cyclophosphamid (64 % vs. 53 %)3).
Steroidhaltige Augentropfen unterdrücken die Symptome einer Episkleritis schnell, es wird jedoch darauf hingewiesen, dass sie nach dem Absetzen einen „Rebound“ mit Rötung und möglicherweise stärkerem Wiederaufflammen verursachen können. Daher ist der Einsatz von Steroiden umstritten; bei leichten Fällen wird teilweise eine abwartende Beobachtung ohne Behandlung oder der Vorzug von NSAIDs empfohlen. Bei wiederholten Rezidiven werden orale COX-2-Hemmer oder eine Abklärung systemischer Erkrankungen empfohlen.
Der Entstehungsmechanismus der Episkleritis ist noch nicht vollständig geklärt. In den betroffenen Bereichen kommt es zu einer Erweiterung und Rötung der oberflächlichen episkleralen Gefäße, mit Infiltration von Entzündungszellen, hauptsächlich Lymphozyten, in die Episklera und die Tenon-Kapsel. Der wesentliche Unterschied zur Skleritis besteht darin, dass das Sklerastroma selbst nicht betroffen ist. Das entzündliche Infiltrat besteht hauptsächlich aus T-Zellen und wenigen Plasmazellen; eine neutrophile eitrige Entzündung oder Granulombildung wird normalerweise nicht beobachtet.
Histopathologisch handelt es sich um eine nicht-granulomatöse Entzündung, die hauptsächlich durch Gefäßerweiterung und lymphozytäre Infiltration gekennzeichnet ist. Bei der nodulären Episkleritis finden sich im Zentrum der Läsion eine fibrinoide Nekrose und eine umgebende Anordnung von Epitheloidzellen. Diese Befunde ähneln dem Bild der granulomatösen Entzündung bei Skleritis, und es gibt die Auffassung, dass Episkleritis und Skleritis als ein Spektrum betrachtet werden können, das sich durch die Tiefe der Entzündung unterscheidet. Die bei der Episkleritis beobachtete kleine fibrinoide Nekrose kann als milde Form der ausgedehnteren nekrotischen Veränderungen bei der Skleritis verstanden werden.
Das Fortschreiten der Entzündung erhöht die Produktion reaktiver Sauerstoffspezies (ROS) und verstärkt den oxidativen Stress2). Der Gesamtvitamin-C-Gehalt der menschlichen Netzhaut ist etwa 20-mal höher als im Plasma, und das Augengewebe ist stark auf das antioxidative System angewiesen. Bei der autoimmunen Episkleritis wird vermutet, dass eine verminderte Funktion dieses antioxidativen Systems zu chronischer Entzündung und Gewebeschädigung der Episklera führen könnte2). ROS schädigen das Gefäßendothel und induzieren die Freisetzung von Entzündungszytokinen, was zu anhaltender Gefäßerweiterung und erhöhter Permeabilität führt. Die chronische Exposition der Augenoberfläche und der Episklera gegenüber oxidativem Stress wird als ein Faktor für die rezidivierende Episkleritis angesehen, und die therapeutische Bedeutung einer antioxidativen Intervention wird untersucht.
Klinisch geht eine Episkleritis selten direkt in eine Skleritis über. Da jedoch bei der Mehrzahl der Skleritisfälle auch eine Entzündung der Episklera (episkleritische Veränderungen) vorliegt, werden beide nicht als völlig unabhängige Erkrankungen, sondern als ein Kontinuum verstanden, das sich durch die Tiefe der betroffenen Gefäßschichten unterscheidet. Die Episkleritis betrifft hauptsächlich das oberflächliche episklerale Gefäßnetz (parietale Schicht), während die Skleritis das tiefe Gefäßnetz und das Sklerastroma befällt.
An der Ansatzstelle der geraden Augenmuskeln ist die Sklera mit etwa 0,3 mm am dünnsten und daher besonders anfällig für Entzündungen und Traumata. Der episklerale Gefäßplexus wird über die vorderen Ziliararterien reichlich mit Blut versorgt, sodass sich bei Entzündungen schnell eine Rötung entwickelt. Die Sklera selbst ist dagegen ein gefäßarmes Gewebe, sodass tiefe Entzündungen wie eine Skleritis selten sind. Die anatomische Besonderheit, dass die von den vorderen Ziliararterien stammenden Gefäße bei Episkleritis reversibel erweitert sind, ist die mechanistische Grundlage dafür, dass die Rötung im Epinephrin-Augentropfentest schnell abklingt; bei tiefer Skleritis tritt diese Reaktion nicht auf, was ein pathophysiologisches Unterscheidungsmerkmal darstellt.
Es gibt einen Fallbericht über einen 60-jährigen Mann mit idiopathischer rezidivierender Episkleritis, bei dem nach Beginn einer oralen Einnahme von Vitamin C 500 mg/Tag über 7 Monate kein Rückfall auftrat 2). Vitamin C ist ein starkes Antioxidans, und es wird darauf hingewiesen, dass es durch die Reduzierung von oxidativem Stress Entzündungen des Augengewebes unterdrücken kann. Es ist bekannt, dass das Augengewebe stark von antioxidativen Systemen abhängig ist, wobei die Vitamin-C-Konzentration in der Netzhaut etwa das 20-fache des Plasmaspiegels erreicht. Eine Supplementierung mit Vitamin C und anderen antioxidativen Nährstoffen könnte daher eine Kandidatenstrategie zur Rückfallprävention darstellen 2). Um die Wirksamkeit zu belegen, sind jedoch kontrollierte Fall-Kontroll-Studien und klinische Studien erforderlich 2). Derzeit wird dies nur als ergänzende Maßnahme bei Fällen mit starken Rückfällen oder bei zugrunde liegendem trockenem Auge oder chronischer Augenoberflächenentzündung in Betracht gezogen.
Die Granulomatose mit Polyangiitis ist unbehandelt eine tödliche Erkrankung mit einer 1-Jahres-Mortalität von 80 %, aber durch die Einführung einer immunsuppressiven Therapie kann die Mortalität auf 10 % gesenkt werden 3). Da eine Episkleritis das erste Symptom einer GPA sein kann, müssen Augenärzte diesen Zusammenhang erkennen und bei rezidivierender Episkleritis aktiv systemische Untersuchungen durchführen 3)4). Insbesondere das gleichzeitige Auftreten von Augenentzündung und Nierenfunktionsstörung ist ein starker Hinweis auf eine Granulomatose mit Polyangiitis 3).
Für Episkleritis und Skleritis im Zusammenhang mit rheumatoider Arthritis wurde die Wirksamkeit von Biologika wie TNFα-Inhibitoren und Rituximab berichtet 1). Infliximab und Adalimumab haben sich bei rheumatoider Arthritis und Uveitis bewährt und werden auch für refraktäre Skleritis und Episkleritis in Betracht gezogen. Andererseits ist bekannt, dass Etanercept paradoxe Reaktionen hervorrufen kann, die Augenentzündungen auslösen oder verschlimmern, sodass bei der Arzneimittelauswahl Vorsicht geboten ist 1). Rituximab ist ein monoklonaler Antikörper, der auf B-Zellen abzielt, und es gibt Hinweise auf seine Wirksamkeit bei vaskulitisbedingten Augenentzündungen. Die Anwendung dieser Biologika wird in enger Zusammenarbeit mit der Rheumatologie und der Abteilung für Kollagenosen entschieden.
Es wurden Fälle berichtet, bei denen Patienten mit der Diagnose einer Episkleritis tatsächlich einen intraokularen metastatischen Tumor 6) oder eine subkonjunktivale Parasitose 7) aufwiesen. Daher ist es bei therapierefraktärer oder rezidivierender Episkleritis wichtig, maligne Erkrankungen oder Infektionen auszuschließen. Bildgebende Untersuchungen und die detaillierte Beurteilung der Spaltlampenbefunde von Gefäße einschließenden Raumforderungen liefern diagnostische Hinweise. Die Beweglichkeit der Raumforderung, die Durchsichtigkeit der Skleragefäße, das Vorhandensein von Verwachsungen mit dem umliegenden Gewebe und das Ansprechen auf die Behandlung müssen gemeinsam beurteilt werden. Anhaltende erhabene Läsionen, die nicht auf die übliche Steroid-Augentropfentherapie ansprechen, sind ein Grund für eine aktive Biopsie und weiterführende Bildgebung.
Langzeitbeobachtungsstudien zum natürlichen Verlauf der Episkleritis und zur Zeit bis zum Auftreten systemischer Erkrankungen sind begrenzt. Insbesondere für die japanische Bevölkerung liegen keine ausreichenden Daten zur Inzidenz und zum Profil der Begleiterkrankungen vor. Bisherige Zusammenstellungen aus westlichen Ländern zeigen eine jährliche Inzidenz von etwa 40–60 Fällen pro 100.000 Personen, wobei die Zahlen aufgrund von Unterschieden in Ethnie, Lebensumfeld und der Führung von Uveitis-Registern variieren. Zukünftige klinische Register und multizentrische Studien werden erwartet, um Risikofaktoren für ein Rezidiv und den zeitlichen Verlauf bis zum Auftreten systemischer Erkrankungen zu identifizieren.
- Promelle V, Goeb V, Gueudry J. Rheumatoid Arthritis Associated Episcleritis and Scleritis: An Update on Treatment Perspectives. Journal of clinical medicine. 2021;10(10). doi:10.3390/jcm10102118. PMID:34068884; PMCID:PMC8156434.
- Goyal L, Ajmera K, Pandit R. Reoccurring Episcleritis and the Role of Antioxidants. Cureus. 2022;14(4):e24111. doi:10.7759/cureus.24111. PMID:35530867; PMCID:PMC9073074.
- Foster LD, Nyugen M, Margolin E. Conjunctivitis, episcleritis and anterior uveitis as the first presenting features of granulomatosis with polyangiitis. BMJ Case Rep. 2021;14:e243558. doi:10.1136/bcr-2021-243558.
- Ciotoracu AC, Dimăncescu MG, Mitulescu TC, et al. A clinical case of recurrent episcleritis as the initial manifestation of granulomatosis with polyangiitis. Rom J Ophthalmol. 2021;65(4):386-390. doi:10.22336/rjo.2021.76.
- Jari M, Nasiri S, Ghandehari M. Episcleritis and posterior uveitis misdiagnosed as orbital cellulitis in a child patient with Behçet’s disease. SAGE Open Med Case Rep. 2023;11:1-4. doi:10.1177/2050313x231182237.
- Chong YJ, Azzopardi M, Ng B, Salvi SM, Sreekantam S. Ocular Metastasis as First Presentation of Large-Cell Neuroendocrine Carcinoma. Case reports in ophthalmology. 2023;14(1):684-691. doi:10.1159/000535233. PMID:38090108; PMCID:PMC10715755.
- Redón-Soriano M, Blasco A, Gomila B, González-Sánchez M, Simón F, Esteban JG. Subconjunctival human dirofilariasis by Dirofilaria repens in the Mediterranean Basin. American journal of ophthalmology case reports. 2022;26:101570. doi:10.1016/j.ajoc.2022.101570. PMID:35586152; PMCID:PMC9108447.