Episclerite simples
Frequência: Mais comum
Início: Súbito
Curso: Atinge o pico em cerca de 12 horas e desaparece em 2 a 3 dias
Achados: Hiperemia em leque (cerca de 67%) ou difusa (cerca de 33%)

A episclerite é uma doença hiperêmica benigna e autolimitada do tecido episcleral. É uma inflamação dos plexos vasculares superficiais, como o plexo da cápsula de Tenon, e, em comparação com a esclerite, que afeta vasos mais profundos, a dor é mais leve e o impacto na visão é menor. Na maioria dos casos, é idiopática e recorrente, com tendência a ocorrer em ambos os olhos. A incidência relatada é de 41,0 casos por 100.000 pessoas por ano, e a prevalência é de 52,6.
Embora seja uma causa relativamente comum de hiperemia, a episclerite é frequentemente confundida com conjuntivite ou esclerite, levando a erros diagnósticos na primeira consulta. Nesta doença, o parênquima escleral não é afetado e a progressão para complicações estruturais graves, como perfuração ocular, é praticamente inexistente. No entanto, em casos com curso recorrente ou naqueles com doenças autoimunes sistêmicas de base, como artrite reumatoide ou granulomatose com poliangeíte, são necessários tratamento da doença de base e acompanhamento de longo prazo. Compreender a episclerite não apenas como uma doença ocular isolada, mas como uma “manifestação ocular” de uma doença sistêmica, está diretamente ligado ao manejo das recidivas e à melhora do prognóstico.
A classificação de Watson é amplamente utilizada para a classificação clínica das doenças inflamatórias da esclera e episclera. Elas são divididas em três grupos principais de acordo com a localização: episclerite, esclerite anterior e esclerite posterior. A esclerite anterior é ainda subdividida em difusa, nodular e necrosante (inflamatória/não inflamatória) com base na morfologia. Uma diferença importante em relação à esclerite anterior é que a episclerite não possui um tipo necrosante e é morfologicamente classificada em dois tipos: simples (difusa) e nodular. Essa classificação reflete a profundidade da inflamação (superficial ou profunda) e a gravidade da progressão e prognóstico, portanto, a determinação do tipo de doença no momento do diagnóstico é a base para a estratégia de tratamento e explicação do prognóstico. A episclerite é classificada como o grupo mais leve e de melhor prognóstico nesta classificação.
Episclerite simples
Frequência: Mais comum
Início: Súbito
Curso: Atinge o pico em cerca de 12 horas e desaparece em 2 a 3 dias
Achados: Hiperemia em leque (cerca de 67%) ou difusa (cerca de 33%)
Episclerite nodular
Frequência: Um pouco menos comum
Início: Insidioso
Curso: Tendência a sintomas mais prolongados do que a episclerite simples
Achados: Nódulo episcleral localizado próximo ao limbo corneano (móvel)
A esclera é composta por três camadas: episclera, estroma escleral e lâmina fusca. A episclera é um tecido conjuntivo vascularizado sobre o estroma escleral, entendida como uma estrutura fibroelástica localizada entre o estroma escleral e a cápsula de Tenon. Consiste em duas camadas: a camada parietal externa (rede capilar episcleral superficial) e a camada visceral profunda (rede vascular altamente anastomosada), ambas derivadas das artérias ciliares anteriores. A maioria das fibras nervosas são ramos do nervo trigêmeo. A episclera forma o plexo vascular episcleral entre a inserção dos músculos retos e o limbo, normalmente escondido pela conjuntiva, mas quando inflamada, dilata-se e produz uma hiperemia vívida. A episclera torna-se gradualmente mais fina em direção à parte posterior do olho, onde a cápsula de Tenon predomina.
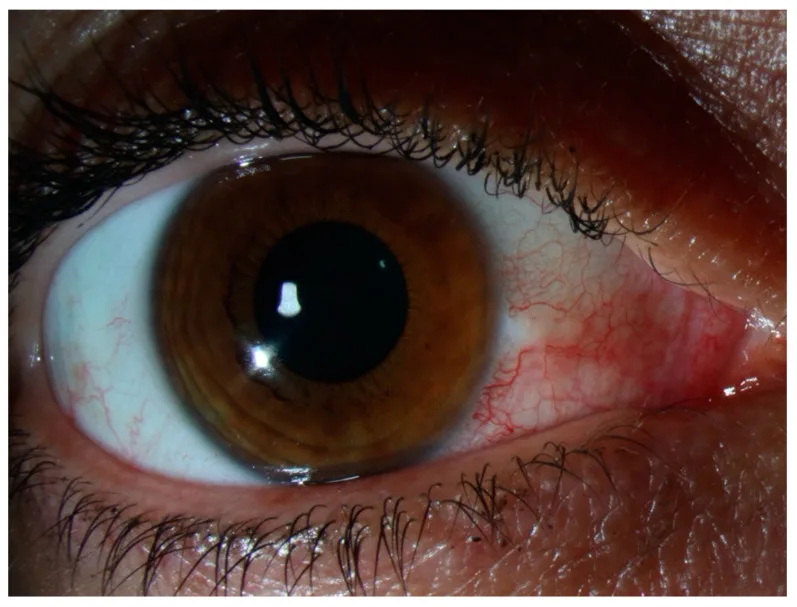
Não há dor à palpação nem secreção ocular. Na presença de dor intensa ou secreção evidente, reconsiderar esclerite, conjuntivite infecciosa ou uveíte anterior. Os sintomas geralmente melhoram ou desaparecem completamente em poucos dias, sem afetar a função visual. Na recorrência, ocorre frequentemente no mesmo local ou no olho contralateral, e o paciente muitas vezes percebe como “o mesmo olho vermelho de sempre”. Dor intensa que interfere no sono noturno, como na esclerite, ou dor forte à palpação da pálpebra superior geralmente não estão presentes na episclerite.
A observação da localização e cor da hiperemia é central para o diagnóstico diferencial. A hiperemia na episclerite é vermelho-vivo a rosada, em contraste com a hiperemia profunda vermelho-escura (arroxeada) da esclerite.
| Achado | Episclerite | Esclerite |
|---|---|---|
| Cor da hiperemia | Vermelho-vivo a rosado | Vermelho-escuro (arroxeado) |
| Dor | Leve a ausente | Forte e irradiada |
| Mobilidade do nódulo | Presente | Ausente |
A acuidade visual é geralmente normal. Edema conjuntival, hipertensão ocular, uveíte anterior e ceratite são complicações raras; quando presentes, considerar esclerite ou outras doenças. A ausência de inflamação na conjuntiva tarsal é útil para diferenciar da conjuntivite. Na esclerite, a inflamação pode se espalhar para tecidos adjacentes, causando infiltrados corneanos periféricos, úlceras ou uveíte anterior, enquanto a episclerite é autolimitada e raramente envolve tecidos vizinhos. Na lâmpada de fenda, identifica-se o nível do plexo vascular escleral; se uma lesão elevada e avermelhada não permitir a visualização dos vasos escleraise, considerar a possibilidade de lesão neoplásica.
A episclerite não apresenta secreção ocular, e a hiperemia é localizada próxima ao limbo corneano. A conjuntivite geralmente é indolor, com secreção ocular, e a hiperemia é mais proeminente no fórnice, diminuindo em direção ao limbo. Na lâmpada de fenda, os vasos episclerais não são móveis, enquanto os vasos conjuntivais são móveis, outro ponto de diferenciação. Consulte a seção “Diagnóstico e Métodos de Exame” para detalhes.
A maioria é idiopática (causa desconhecida), com relatos de associação com doenças sistêmicas em cerca de 26 a 36% dos casos. Mesmo nos casos idiopáticos, sugere-se envolvimento de mecanismos imunológicos, com base em uma reação inflamatória inespecífica centrada em linfócitos no plexo vascular episcleral superficial. O curso recorrente e a tendência bilateral sugerem uma desregulação imunológica sistêmica subjacente.
Doenças do colágeno e autoimunes (a mais comum é a artrite reumatoide)1):
Vasculite:
Infecções: bactérias, micobactérias, sífilis, doença de Lyme, vírus herpes, herpes zóster, entre outros, podem ser causas. A episclerite associada ao herpes zóster oftálmico é considerada uma resposta imune ao patógeno, e não a infecção em si. Também foi relatado um caso de parasitose subconjuntival por Dirofilaria repens diagnosticada erroneamente como episclerite7).
Outros: gota, atopia, corpo estranho, trauma químico, medicamentos (topiramato, pamidronato) e relatos como sintoma inicial da COVID-19.
Sim, cerca de 30% dos pacientes apresentam doença sistêmica associada. A mais comum é a artrite reumatoide, mas também pode ser o primeiro sintoma de doenças como granulomatose com poliangiite (GPA) ou doença de Behçet, cujo diagnóstico e tratamento precoces afetam o prognóstico. Em casos de recorrência frequente ou sintomas sistêmicos, recomenda-se uma investigação sistêmica incluindo fator reumatoide, anticorpos antinucleares, ANCA e exame de urina.
A episclerite é diagnosticada principalmente por meio de anamnese e exame com lâmpada de fenda. O básico é observar cuidadosamente com a lâmpada de fenda o nível dos vasos escleral (superficial ou profundo), a tonalidade da hiperemia, a presença de nódulos e a presença de afinamento ou necrose.
O colírio de fenilefrina a 2,5% contrai os vasos conjuntivais e é útil para diferenciar conjuntivite de episclerite. O colírio de fenilefrina a 10% contrai a rede vascular escleral superficial, mas não a profunda, permitindo diferenciar episclerite de esclerite.
O teste de reação ao colírio de epinefrina diluído 1:1000 é um método simples para avaliar o envolvimento de vasos profundos. Se a hiperemia desaparecer após a instilação, sugere episclerite; se não desaparecer, sugere esclerite. A avaliação é feita combinando três pontos: número e mobilidade dos nódulos, presença de dor/ sensibilidade à palpação e reação à epinefrina.
Os testes de reação com epinefrina e fenilefrina são particularmente úteis como auxílio diagnóstico quando a estrutura em camadas da hiperemia não pode ser confirmada diretamente com a lâmpada de fenda ou em casos com nódulos pequenos. A observação 10 a 15 minutos após a instilação determina a presença ou ausência de contração dos vasos superficiais; se a hiperemia dos vasos profundos persistir, prioriza-se o manejo para esclerite.
A tenonite também é considerada um tipo de episclerite, sendo difícil a diferenciação clínica entre ambas. A avaliação combina a mobilidade do nódulo, presença de dor e dor à palpação, resposta à instilação de epinefrina e achados da coloração com fluoresceína.
Na episclerite única e leve, não é necessária uma investigação sistêmica ampla. Em casos de recorrência ou com sintomas sistêmicos, considerar os seguintes exames.
Em casos de episclerite como manifestação inicial de granulomatose com poliangeíte, pode haver comprometimento renal concomitante3). Quando tanto a inflamação ocular quanto a disfunção renal estão presentes, deve-se realizar prontamente a investigação de vasculite sistêmica, incluindo granulomatose com poliangeíte. Na episclerite refratária ou recorrente, é desejável avaliar a atividade da doença e iniciar o tratamento da doença de base em colaboração com a reumatologia e a clínica médica.
Além da avaliação com lâmpada de fenda, a tomografia de coerência óptica do segmento anterior (AS-OCT) para avaliar a espessura da camada episcleral e o trajeto vascular, e a ultrassonografia (modo B) para avaliar a espessura escleral podem ser usadas como exames auxiliares. Para excluir esclerite necrosante ou avaliar a presença de esclerite posterior, a ultrassonografia verifica a presença do sinal T (acúmulo de líquido ao redor da bainha do nervo óptico). Na episclerite comum, esses exames de imagem frequentemente não apresentam achados específicos, e o diagnóstico é feito pela combinação do exame direto com lâmpada de fenda, anamnese e investigação sistêmica.
A epiesclerite geralmente cura espontaneamente em dias a semanas sem tratamento. Explicar ao paciente a natureza benigna da doença, seu curso natural e a necessidade de investigar doenças sistêmicas, além de proporcionar tranquilidade, é o primeiro passo no manejo. Compressas frias e lágrimas artificiais geladas são eficazes para reduzir sintomas subjetivos como irritação e sensação de calor. Em casos leves, a intervenção medicamentosa agressiva não é realizada, e o acompanhamento de curto prazo (em dias) para confirmar a melhora espontânea evita rebotes e efeitos colaterais associados ao tratamento.
Colírios de corticosteroides de baixa concentração são a primeira escolha. Muitas vezes, também são usados colírios antibióticos em conjunto para auxiliar na diferenciação da esclerite.
Se a resposta ao tratamento com colírios for insuficiente, considere mudar para exames e tratamento de esclerite. Embora os colírios de esteroides aliviem rapidamente os sintomas, o uso prolongado e repetido pode aumentar o risco de recorrência e induzir hiperemia de “rebote”.
O tratamento baseia-se na redução gradual e descontinuação após o desaparecimento dos sintomas, evitando a administração contínua e indiscriminada. O uso prolongado de colírios de corticosteroides apresenta risco de aumento da pressão intraocular responsivo a esteroides e catarata subcapsular posterior, portanto, a redução gradual deve ser feita após confirmar melhora em 1 a 2 semanas. Em casos de recorrência, avalie individualmente a atividade da doença em cada episódio e priorize a otimização do tratamento da doença sistêmica subjacente.
Na esclerite superior associada a doenças do colágeno, como artrite reumatoide, o tratamento da doença de base está diretamente ligado ao prognóstico 1). Caso o tratamento local seja resistente, utiliza-se prednisolona oral (terapia de redução gradual a partir de 20-30 mg/dia). Exceto quando há evidência clara de doença inflamatória sistêmica concomitante, os casos que necessitam de corticoterapia sistêmica são muito raros.
Na episclerite associada à granulomatose com poliangiite, a terapia de indução de remissão com ciclofosfamida ou rituximabe é eficaz3)4). Há relatos de que o rituximabe apresenta maior taxa de remissão em 6 meses em comparação com a ciclofosfamida (64% vs. 53%)3).
Colírios de esteroides suprimem rapidamente os sintomas da episclerite, mas foi apontado que podem causar hiperemia de “rebote” após a descontinuação, levando a uma recidiva mais intensa. Portanto, o uso de esteroides é controverso, e em casos leves, há opiniões que priorizam a observação sem tratamento ou AINEs. Em casos de recorrência, recomenda-se o uso oral de inibidores da COX-2 e a investigação de doenças sistêmicas.
O mecanismo de início da episclerite ainda não foi completamente elucidado. Na área afetada, ocorrem dilatação vascular e congestão na rede vascular episcleral superficial, com infiltração de células inflamatórias, principalmente linfócitos, na episclera e na cápsula de Tenon. A diferença essencial da esclerite é que o parênquima escleral em si não é afetado. O infiltrado inflamatório consiste principalmente em células T e alguns plasmócitos, e geralmente não se observa inflamação purulenta com predominância de neutrófilos ou formação de granuloma.
Histopatologicamente, é uma inflamação não granulomatosa, com dilatação vascular e infiltração linfocitária predominantes. Na episclerite nodular, observam-se necrose fibrinóide no centro da lesão e um arranjo de células epitelióides ao redor. Esses achados são semelhantes aos da inflamação granulomatosa observada na esclerite, e há a visão de que a episclerite e a esclerite formam um espectro com base na profundidade da inflamação. A necrose fibrinóide de pequena escala observada na episclerite pode ser entendida como a extremidade leve das alterações necróticas mais extensas na esclerite.
A progressão da inflamação aumenta a produção de espécies reativas de oxigênio (ROS) e intensifica o estresse oxidativo2). A quantidade total de vitamina C na retina humana é cerca de 20 vezes maior que no plasma, e o tecido ocular depende fortemente do sistema antioxidante. Na episclerite autoimune, sugere-se que a disfunção desse sistema antioxidante pode causar inflamação crônica e dano tecidual na episclera2). As ROS danificam o endotélio vascular e induzem a liberação de citocinas inflamatórias, causando dilatação vascular persistente e aumento da permeabilidade. A exposição crônica ao estresse oxidativo na superfície ocular e na episclera tem sido apontada como um fator contribuinte para a episclerite recorrente, e o significado terapêutico da intervenção antioxidante está sendo investigado.
Clinicamente, a episclerite raramente transita diretamente para esclerite. Por outro lado, como na maioria dos casos de esclerite também há inflamação na episclera (alterações episcleríticas), ambas são entendidas não como doenças completamente independentes, mas como um continuum baseado na profundidade da camada vascular afetada pela inflamação. A episclerite afeta principalmente a rede vascular episcleral superficial (camada parietal), enquanto a esclerite afeta desde a rede vascular profunda até o parênquima escleral.
Na inserção dos músculos retos, a espessura da esclera é de cerca de 0,3 mm, a mais fina, sendo conhecida por sua alta vulnerabilidade a inflamação e trauma. O plexo vascular episcleral recebe suprimento sanguíneo abundante através das artérias ciliares anteriores, de modo que a congestão tende a se manifestar rapidamente durante a inflamação. Por outro lado, a esclera em si é um tecido pobre em vasos, e a inflamação profunda como a esclerite é rara. A característica anatômica de que os vasos derivados das artérias ciliares anteriores na episclerite se congestionam reversivelmente é a base mecanicista para a rápida resolução da congestão no teste com colírio de epinefrina, e a ausência dessa reação na vasculite escleral profunda serve como base fisiopatológica para o diagnóstico diferencial.
Há um relato de caso de um homem de 60 anos com episclerite idiopática recidivante que, após iniciar vitamina C 500 mg/dia por via oral, não apresentou recidiva por 7 meses2). A vitamina C é um potente antioxidante e acredita-se que possa suprimir a inflamação do tecido ocular através da redução do estresse oxidativo. Sabe-se que o tecido ocular tem alta dependência do sistema antioxidante, com a concentração de vitamina C na retina atingindo cerca de 20 vezes a do plasma, e a suplementação de vitamina C e outros nutrientes antioxidantes pode ser uma candidata a estratégia de prevenção de recidiva2). No entanto, para estabelecer a eficácia, são necessários estudos caso-controle com grupo controle e ensaios clínicos no futuro2). Atualmente, considera-se apenas como adjuvante em casos de recidiva com sintomas intensos ou com olho seco e inflamação crônica da superfície ocular subjacente.
A granulomatose com poliangiite é uma doença fatal que, se não tratada, tem taxa de mortalidade de 80% em um ano, mas com terapia imunossupressora a mortalidade pode ser reduzida para 10%3). Como a episclerite pode ser o primeiro sintoma da GPA, os oftalmologistas devem reconhecer essa associação e realizar exames sistêmicos ativamente em casos de episclerite recidivante3)4). Especialmente a coexistência de inflamação ocular e disfunção renal é um achado que sugere fortemente granulomatose com poliangiite3).
A eficácia de agentes biológicos como inibidores de TNFα e rituximabe foi relatada para episclerite e esclerite associadas à artrite reumatoide1). Infliximabe e adalimumabe têm histórico no tratamento de artrite reumatoide e uveíte, e são considerados para esclerite e episclerite refratárias. Por outro lado, o etanercepte é conhecido por induzir ou exacerbar inflamação ocular como uma reação paradoxal, exigindo cautela na seleção do medicamento1). O rituximabe é um anticorpo monoclonal direcionado às células B, e sua eficácia para inflamação ocular relacionada a vasculite tem sido sugerida. O uso desses agentes biológicos é decidido em estreita colaboração com reumatologistas e especialistas em doenças do colágeno.
Foram relatados casos em que pacientes diagnosticados com episclerite na verdade apresentavam tumor metastático intraocular 6) ou parasitose subconjuntival 7), sendo importante excluir doenças malignas ou infecciosas na episclerite refratária e recidivante. Exames de imagem e avaliação detalhada da massa com vasos à lâmpada de fenda são pistas diagnósticas. A mobilidade da massa, a transparência dos vasos esclerais, a presença de aderências aos tecidos circundantes e a resposta ao tratamento devem ser avaliados em conjunto; lesões elevadas persistentes que não respondem ao colírio de esteroide usual justificam consideração ativa de biópsia e exames de imagem detalhados.
Estudos de observação de longo prazo sobre a história natural da episclerite e o período até a manifestação de doenças sistêmicas são limitados, e os dados sobre a incidência e o perfil de doenças associadas na população japonesa são insuficientes. Relatos anteriores de países ocidentais mostram uma incidência anual de aproximadamente 40 a 60 casos por 100.000 pessoas, mas os números variam devido a diferenças étnicas, ambientais e na operação de registros de uveíte. Espera-se que a construção futura de registros clínicos e estudos multicêntricos identifiquem fatores de risco de recidiva e o cronograma até a manifestação de doenças sistêmicas.
- Promelle V, Goeb V, Gueudry J. Rheumatoid Arthritis Associated Episcleritis and Scleritis: An Update on Treatment Perspectives. Journal of clinical medicine. 2021;10(10). doi:10.3390/jcm10102118. PMID:34068884; PMCID:PMC8156434.
- Goyal L, Ajmera K, Pandit R. Reoccurring Episcleritis and the Role of Antioxidants. Cureus. 2022;14(4):e24111. doi:10.7759/cureus.24111. PMID:35530867; PMCID:PMC9073074.
- Foster LD, Nyugen M, Margolin E. Conjunctivitis, episcleritis and anterior uveitis as the first presenting features of granulomatosis with polyangiitis. BMJ Case Rep. 2021;14:e243558. doi:10.1136/bcr-2021-243558.
- Ciotoracu AC, Dimăncescu MG, Mitulescu TC, et al. A clinical case of recurrent episcleritis as the initial manifestation of granulomatosis with polyangiitis. Rom J Ophthalmol. 2021;65(4):386-390. doi:10.22336/rjo.2021.76.
- Jari M, Nasiri S, Ghandehari M. Episcleritis and posterior uveitis misdiagnosed as orbital cellulitis in a child patient with Behçet’s disease. SAGE Open Med Case Rep. 2023;11:1-4. doi:10.1177/2050313x231182237.
- Chong YJ, Azzopardi M, Ng B, Salvi SM, Sreekantam S. Ocular Metastasis as First Presentation of Large-Cell Neuroendocrine Carcinoma. Case reports in ophthalmology. 2023;14(1):684-691. doi:10.1159/000535233. PMID:38090108; PMCID:PMC10715755.
- Redón-Soriano M, Blasco A, Gomila B, González-Sánchez M, Simón F, Esteban JG. Subconjunctival human dirofilariasis by Dirofilaria repens in the Mediterranean Basin. American journal of ophthalmology case reports. 2022;26:101570. doi:10.1016/j.ajoc.2022.101570. PMID:35586152; PMCID:PMC9108447.