Simple episcleritis
Frequency: More common
Onset: Acute
Course: Peaks in about 12 hours, resolves in 2–3 days
Findings: Sectoral (about 67%) or diffuse (about 33%) hyperemia

Episcleritis is a benign, self-limited congestive condition of the episcleral tissue. It involves inflammation of the superficial vascular plexus, such as the Tenon’s capsule vascular plexus, and is less painful and has less impact on vision compared to scleritis, which affects deeper vessels. Most cases are idiopathic and recurrent, with a tendency for bilateral involvement. The reported incidence is 41.0 per 100,000 person-years, and the prevalence is 52.6 per 100,000.
While it is a relatively common cause of conjunctival injection, it can be confused with conjunctivitis or scleritis, leading to misdiagnosis at initial presentation. In this condition, the scleral stroma itself is not affected, and progression to severe structural complications such as globe perforation is rare. However, in cases with a recurrent course or those associated with systemic autoimmune diseases such as rheumatoid arthritis or granulomatosis with polyangiitis, treatment of the underlying disease and long-term follow-up are necessary. Understanding episcleritis not merely as an isolated ocular disease but as an ocular manifestation of systemic disease is crucial for recurrence management and prognosis improvement.
The Watson classification is widely used for the clinical classification of inflammatory diseases of the sclera and episclera. It broadly divides them into three groups by site: episcleritis, anterior scleritis, and posterior scleritis. Anterior scleritis is further subdivided by morphology into diffuse, nodular, and necrotizing (inflammatory/non-inflammatory) types. An important difference from anterior scleritis is that episcleritis has no necrotizing type and is morphologically classified into two types: simple (diffuse) and nodular. Since this classification reflects the depth of inflammation (superficial or deep) and the severity of progression and prognosis, determining the disease type at diagnosis forms the basis for treatment strategy and prognosis explanation. Episcleritis is positioned as the mildest and most prognostically favorable group in this classification.
Simple episcleritis
Frequency: More common
Onset: Acute
Course: Peaks in about 12 hours, resolves in 2–3 days
Findings: Sectoral (about 67%) or diffuse (about 33%) hyperemia
Nodular episcleritis
Frequency: Slightly less common
Onset: Gradual
Course: Tends to be more prolonged than simple type
Findings: Localized episcleral nodule near the limbus (mobile)
The sclera consists of three layers: the episclera, scleral stroma, and lamina fusca. The episclera is a connective tissue containing blood vessels overlying the scleral stroma, understood as a fibroelastic structure located between the scleral stroma and Tenon’s capsule. It comprises two layers: an outer parietal layer (superficial episcleral capillary network) and a deep visceral layer (highly anastomotic vascular network), both derived from the anterior ciliary arteries. Most nerve fibers are branches of the trigeminal nerve. The episclera forms the episcleral vascular plexus between the rectus muscle insertions and the limbus, usually hidden by the conjunctiva, but when inflamed, it dilates and produces bright hyperemia. The episclera gradually becomes thinner toward the posterior part of the eye, where Tenon’s capsule predominates.
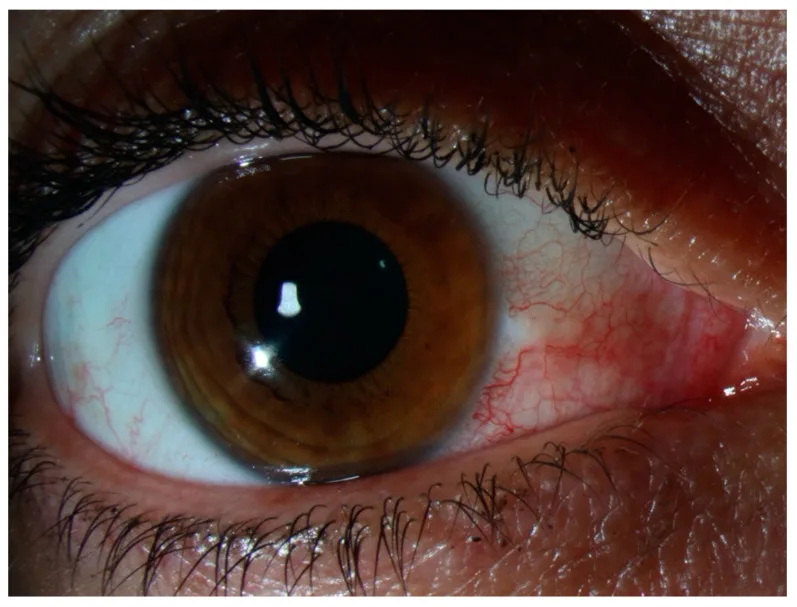
There is no tenderness and no discharge. If severe pain or obvious discharge is present, reconsider scleritis, infectious conjunctivitis, or anterior uveitis. Symptoms often improve or resolve completely within a few days, leaving no impact on visual function. Recurrences often occur in the same location or the contralateral eye, and patients often recognize it as “the usual red eye.” Severe pain that disturbs nighttime sleep, as in scleritis, or strong tenderness when touching the upper eyelid is usually not seen in episcleritis.
Observation of the location and color of injection is central to differential diagnosis. The injection in episcleritis is bright red to pink, in contrast to the dark red (violaceous) deep injection seen in scleritis.
| Finding | Episcleritis | Scleritis |
|---|---|---|
| Color of injection | Bright red to pink | Dark red (violaceous) |
| Pain | Mild to none | Severe, radiating |
| Nodule mobility | Present | Absent |
Visual acuity is generally normal. Conjunctival chemosis, elevated intraocular pressure, anterior uveitis, and keratitis are rare complications; if present, consider scleritis or other diseases. The absence of inflammatory findings in the palpebral conjunctiva is useful for differentiating from conjunctivitis. In scleritis, inflammation may spread to adjacent tissues, causing peripheral corneal infiltrates, ulcers, or anterior uveitis, whereas episcleritis is self-limiting and almost never involves adjacent tissues. On slit-lamp examination, identify the level of the scleral vascular plexus; if a red elevated lesion is present but scleral vessels are not visible, consider the possibility of a neoplastic lesion.
Episcleritis is characterized by the absence of discharge and hyperemia localized near the corneal limbus. Conjunctivitis is usually painless with discharge, and hyperemia is most prominent in the fornix, diminishing toward the limbus. On slit-lamp examination, episcleral vessels are immobile, while conjunctival vessels are mobile, which is another distinguishing feature. For details, see the section on “Diagnosis and Testing Methods”.
Most cases are idiopathic (cause unknown), with systemic disease reported in approximately 26–36% of all cases. Even in idiopathic cases, an immunological mechanism is suggested, based on a nonspecific inflammatory reaction centered on lymphocytes in the superficial episcleral vascular plexus. The recurrent course and tendency for bilateral involvement suggest an underlying systemic immune dysregulation.
Collagen diseases/autoimmune diseases (most commonly rheumatoid arthritis)1):
Vasculitis:
Infections: Bacteria, mycobacteria, syphilis, Lyme disease, herpes virus, herpes zoster, etc. can be causes. Episcleritis associated with ophthalmic herpes zoster is thought to be an immune response to the pathogen rather than the infection itself. There is also a report of a subconjunctival parasitic infection caused by Dirofilaria repens being misdiagnosed as episcleritis7).
Others: Gout, atopy, foreign bodies, chemical trauma, medications (topiramate, pamidronate), and reports as an initial symptom of COVID-19.
Yes, about 30% of patients have an associated systemic disease. The most common is rheumatoid arthritis, but it can also be the initial symptom of diseases where early diagnosis and treatment affect prognosis, such as granulomatosis with polyangiitis (GPA) or Behçet’s disease. In cases of recurrent episodes or accompanying systemic symptoms, systemic workup including rheumatoid factor, antinuclear antibody, ANCA, and urinalysis is recommended.
Episcleritis is primarily a clinical diagnosis based on history taking and slit-lamp examination. The basics involve carefully observing with a slit lamp the level of scleral vessels (superficial or deep), the color of congestion, presence of nodules, and thinning or necrosis.
2.5% phenylephrine eye drops constrict conjunctival vessels and are useful for differentiating conjunctivitis from episcleritis. 10% phenylephrine eye drops constrict the superficial episcleral vascular network but not the deep network, allowing differentiation between episcleritis and scleritis.
The response test using 1:1000 diluted epinephrine eye drops is a simple method to determine deep vascular involvement. If congestion subsides after instillation, it suggests episcleritis; if not, it suggests scleritis. A comprehensive evaluation is made by combining three factors: number and mobility of nodules, presence of pain/tenderness, and epinephrine response.
Response tests with epinephrine and phenylephrine are particularly useful as adjunctive diagnostics when the layered structure of congestion cannot be directly confirmed with a slit lamp or in cases with small nodules. Observation 10–15 minutes after instillation determines the presence or absence of superficial vessel constriction; if deep vessel congestion remains, priority is given to managing scleritis.
Tenonitis is also considered a type of episcleritis, and clinical differentiation between the two is difficult. Judgment is made by combining findings of nodule mobility, presence of pain/tenderness, response to epinephrine eye drops, and fluorescein staining.
For a single, mild episode of episcleritis, extensive systemic workup is unnecessary. In cases of recurrence or when accompanied by systemic symptoms, consider the following tests.
In cases where episcleritis appears as the initial manifestation of granulomatosis with polyangiitis, renal dysfunction may coexist 3). When both ocular inflammation and renal function abnormalities are present, promptly investigate systemic vasculitis including granulomatosis with polyangiitis. For refractory or recurrent episcleritis, it is desirable to evaluate disease activity and initiate treatment for the underlying disease in collaboration with rheumatology and internal medicine.
In addition to slit-lamp microscopy, anterior segment optical coherence tomography (AS-OCT) to assess episcleral thickness and vascular patterns, and ultrasound (B-mode) to evaluate scleral thickness may be used as ancillary diagnostic tools. To rule out necrotizing scleritis or assess posterior scleritis, ultrasound can check for the T-sign (fluid accumulation around the optic nerve sheath). In typical episcleritis, these imaging studies often lack specific findings, and diagnosis is made by combining direct slit-lamp examination with history and systemic evaluation.
Most cases of episcleritis resolve spontaneously within days to weeks without treatment. The first step in management is to explain the benign nature and natural course of the disease to the patient, along with the need for systemic evaluation, and to provide reassurance. Cold compresses and cooled artificial tears are effective in reducing subjective symptoms such as irritation and warmth. In mild cases, avoiding active drug intervention and confirming spontaneous improvement through short-term follow-up over several days can prevent rebound and side effects associated with treatment.
Low-concentration topical steroids are the first-line treatment. Antibiotic eye drops are often used concurrently to help differentiate from scleritis.
If the response to topical treatment is poor, consider switching to evaluation and treatment for scleritis. While topical steroids rapidly suppress symptoms, long-term or repeated use may increase the risk of recurrence and induce “rebound” hyperemia.
Treatment is generally tapered and discontinued after symptom resolution; avoid prolonged continuous use. Long-term use of topical steroids carries risks of steroid-induced intraocular pressure elevation and posterior subcapsular cataract, so improvement should be confirmed within 1–2 weeks before tapering. In recurrent cases, individually assess disease activity for each episode and prioritize optimization of treatment for the underlying systemic disease.
In episcleritis associated with collagen diseases such as rheumatoid arthritis, treatment of the underlying disease directly affects prognosis 1). If local treatment is ineffective, add oral prednisolone (tapering from 20–30 mg/day). Cases requiring systemic steroids are very rare unless there is clear evidence of systemic inflammatory disease.
In episcleritis associated with granulomatosis with polyangiitis, remission induction therapy with cyclophosphamide or rituximab is effective 3)4). Rituximab has been reported to have a higher remission rate at 6 months compared to cyclophosphamide (64% vs. 53%) 3).
Steroid eye drops quickly suppress symptoms of episcleritis, but discontinuation may cause “rebound” redness and potentially lead to stronger flare-ups. Therefore, the use of steroids is debated; for mild cases, some recommend observation without treatment or prioritizing NSAIDs. In recurrent cases, oral COX2 inhibitors or investigation of systemic disease is recommended.
The mechanism of episcleritis onset is not yet fully understood. In affected areas, dilation and congestion occur in the superficial episcleral vascular network, with infiltration of inflammatory cells, mainly lymphocytes, into the episclera and Tenon’s capsule. The essential difference from scleritis is that the scleral stroma itself is not involved. The inflammatory cell infiltrate consists primarily of T cells and a few plasma cells; neutrophil-predominant purulent inflammation or granuloma formation is usually absent.
Histopathologically, it is non-granulomatous inflammation, mainly characterized by vasodilation and lymphocytic infiltration. In nodular episcleritis, fibrinoid necrosis is observed at the center of the lesion, surrounded by a palisade of epithelioid cells. These findings resemble the granulomatous inflammation seen in scleritis, and some views consider episcleritis and scleritis as a spectrum based on the depth of inflammation. The small-scale fibrinoid necrosis observed in episcleritis can be understood as a mild form of the more extensive necrotic changes in scleritis.
Inflammation progression increases the production of reactive oxygen species (ROS), enhancing oxidative stress2). The total vitamin C content in the human retina is about 20 times higher than in plasma, and ocular tissues heavily rely on antioxidant systems. In autoimmune episcleritis, it has been suggested that dysfunction of this antioxidant system may lead to chronic inflammation and tissue damage in the episclera2). ROS damage vascular endothelium and induce the release of inflammatory cytokines, causing persistent vasodilation and increased permeability. Chronic exposure of the ocular surface and episclera to oxidative stress is noted as a contributing factor to recurrent episcleritis, and the therapeutic potential of antioxidant intervention is being investigated.
Clinically, episcleritis rarely transitions directly to scleritis. However, since most cases of scleritis also show inflammation in the episclera (episcleritis-like changes), the two are understood as a continuum based on the depth of vascular layer involvement rather than completely independent diseases. Episcleritis primarily affects the superficial episcleral vascular network (parietal layer), while scleritis involves the deep vascular network and scleral stroma.
At the rectus muscle insertion, the sclera is thinnest at about 0.3 mm, known to be more vulnerable to inflammation and trauma. The episcleral vascular plexus receives abundant blood supply via the anterior ciliary arteries, so congestion becomes rapidly apparent during inflammation. In contrast, the sclera itself is a tissue with sparse blood vessels, making deep inflammation like scleritis rare. The anatomical feature that reversible congestion of anterior ciliary artery-derived vessels occurs in episcleritis is the mechanistic basis for the rapid resolution of congestion with the epinephrine eye drop test; this reaction is absent in deep scleral vasculitis, providing a pathophysiological basis for differentiation.
There is a case report of a 60-year-old man with idiopathic recurrent episcleritis who had no recurrence for 7 months after starting oral vitamin C 500 mg/day 2). Vitamin C is a potent antioxidant, and it has been suggested that it may suppress inflammation in ocular tissues by reducing oxidative stress. Ocular tissues are known to be highly dependent on the antioxidant system, with retinal vitamin C concentrations reaching about 20 times those in plasma, and supplementation with vitamin C and other antioxidant nutrients may be a candidate strategy for recurrence prevention 2). However, controlled case-control studies and clinical trials are needed to establish efficacy 2). At present, it remains at the stage of being considered adjunctively in cases with severe recurrent symptoms or underlying dry eye and chronic ocular surface inflammation.
Granulomatosis with polyangiitis is a fatal disease with a 1-year mortality rate of up to 80% if untreated, but immunosuppressive therapy can reduce mortality to 10% 3). Since episcleritis can be the initial symptom of GPA, ophthalmologists should recognize this association and actively perform systemic evaluation in recurrent episcleritis 3)4). In particular, the coexistence of ocular inflammation and renal dysfunction is a finding strongly suggestive of granulomatosis with polyangiitis 3).
The efficacy of biologic agents such as TNFα inhibitors and rituximab has been reported for episcleritis and scleritis associated with rheumatoid arthritis 1). Infliximab and adalimumab have a track record in rheumatoid arthritis and uveitis, and their application is considered for refractory scleritis and episcleritis. On the other hand, etanercept is known to cause a paradoxical reaction that induces or exacerbates ocular inflammation, requiring caution in drug selection 1). Rituximab is a monoclonal antibody targeting B cells, and its efficacy for vasculitis-related ocular inflammation has been suggested. The use of these biologic agents is determined in close collaboration with rheumatology and collagen disease departments.
Cases have been reported where patients diagnosed with episcleritis actually had intraocular metastatic tumors 6) or subconjunctival parasitic infections 7), highlighting the importance of excluding malignant diseases and infections in refractory or recurrent episcleritis. Imaging studies and detailed slit-lamp evaluation of vascularized masses provide diagnostic clues. Assessment should integrate mass mobility, visibility of scleral vessels, adhesion to surrounding tissues, and treatment response. Persistent elevated lesions unresponsive to standard steroid eye drops warrant consideration of biopsy and advanced imaging.
Long-term observational studies on the natural course of episcleritis and the time to manifestation of systemic diseases are limited, and data on incidence rates and comorbidity profiles in Japanese populations are insufficient. Previous reports from Western countries indicate an annual incidence of approximately 40–60 cases per 100,000 people, but figures vary due to differences in ethnicity, living environment, and uveitis registry operations. Future establishment of clinical registries and multicenter collaborative studies are expected to identify risk factors for recurrence and timelines for systemic disease manifestation.
- Promelle V, Goeb V, Gueudry J. Rheumatoid Arthritis Associated Episcleritis and Scleritis: An Update on Treatment Perspectives. Journal of clinical medicine. 2021;10(10). doi:10.3390/jcm10102118. PMID:34068884; PMCID:PMC8156434.
- Goyal L, Ajmera K, Pandit R. Reoccurring Episcleritis and the Role of Antioxidants. Cureus. 2022;14(4):e24111. doi:10.7759/cureus.24111. PMID:35530867; PMCID:PMC9073074.
- Foster LD, Nyugen M, Margolin E. Conjunctivitis, episcleritis and anterior uveitis as the first presenting features of granulomatosis with polyangiitis. BMJ Case Rep. 2021;14:e243558. doi:10.1136/bcr-2021-243558.
- Ciotoracu AC, Dimăncescu MG, Mitulescu TC, et al. A clinical case of recurrent episcleritis as the initial manifestation of granulomatosis with polyangiitis. Rom J Ophthalmol. 2021;65(4):386-390. doi:10.22336/rjo.2021.76.
- Jari M, Nasiri S, Ghandehari M. Episcleritis and posterior uveitis misdiagnosed as orbital cellulitis in a child patient with Behçet’s disease. SAGE Open Med Case Rep. 2023;11:1-4. doi:10.1177/2050313x231182237.
- Chong YJ, Azzopardi M, Ng B, Salvi SM, Sreekantam S. Ocular Metastasis as First Presentation of Large-Cell Neuroendocrine Carcinoma. Case reports in ophthalmology. 2023;14(1):684-691. doi:10.1159/000535233. PMID:38090108; PMCID:PMC10715755.
- Redón-Soriano M, Blasco A, Gomila B, González-Sánchez M, Simón F, Esteban JG. Subconjunctival human dirofilariasis by Dirofilaria repens in the Mediterranean Basin. American journal of ophthalmology case reports. 2022;26:101570. doi:10.1016/j.ajoc.2022.101570. PMID:35586152; PMCID:PMC9108447.