Systemic lupus erythematosus (SLE) is an autoimmune disease that causes inflammation in multiple organs throughout the body due to chronic overactivation of the immune system. It is considered one of the classic five collagen diseases.
SLE predominantly affects women in their 20s to 30s. The male-to-female ratio is approximately 1:8 to 1:9, with a strong female predominance. It is more common in Asian populations and less common in Caucasians. The prevalence is estimated to be 50–100 per 100,000 population.
The diagnostic criteria for SLE have evolved as follows.
American College of Rheumatology (ACR) criteria (final revision 1997): Diagnosis requires 4 or more of 11 clinical and immunological items.
SLICC criteria (2012): Expanded to 17 items, diagnosis requires 4 or more items.
European League Against Rheumatism/American College of Rheumatology criteria (2019): Positive antinuclear antibody (ANA) is mandatory, diagnosis requires a weighted score of 10 or more.
According to the revised diagnostic criteria of the American College of Rheumatology, diagnosis is possible if 4 or more of the following 11 items are positive during the course: malar rash, discoid rash, photosensitivity, oral ulcers, arthritis, serositis, renal disorder, neurological disorder, hematologic disorder, immunologic disorder, and antinuclear antibody.
In the uveitis clinical practice guidelines, SLE is considered a differential diagnosis for uveitis associated with collagen disease 1). Ocular findings and systemic symptoms are combined and evaluated in collaboration with a rheumatology department.
QAre ocular symptoms of SLE included in the diagnostic criteria?
A
No. Although ocular symptoms occur in about 30% of patients, ocular symptoms themselves are not included in the diagnostic criteria for SLE.
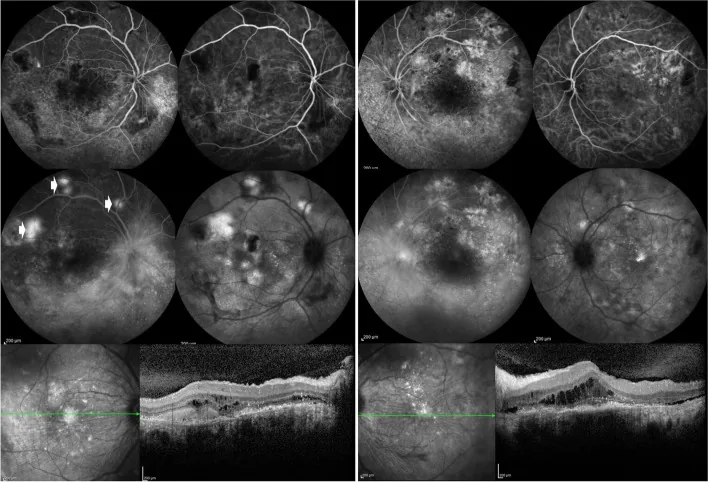
Özdal PÇ, et al. Choroidal involvement in systemic vasculitis: a systematic review. J Ophthalmic Inflamm Infect. 2022. Figure 5. PMCID: PMC8980189. License: CC BY.
Fluorescein angiography of both eyes shows leakage from the optic disc and around retinal vessels, and patchy lesions in the posterior pole. OCT shows subretinal fluid, indicating posterior segment inflammatory lesions associated with SLE.
Ocular complaints are observed in approximately 33–50% of patients. Symptoms range from mild irritation to severe vision loss.
Dryness and foreign body sensation: The most common complaint. Caused by dry eye due to secondary Sjögren’s syndrome. Accompanied by burning sensation, blurred vision, and worsening of symptoms in the evening.
Decreased visual acuity: Occurs in lupus retinopathy and optic neuritis. Ranges from asymptomatic fundus changes to sudden vision loss.
Eye pain: In optic neuritis, pain around the orbit that worsens with eye movement is characteristic.
Ocular motor disturbances: The oculomotor and abducens nerves are affected by central nervous system lesions. Seen in about 30%.
QDoes SLE cause uveitis?
A
Complication with uveitis is surprisingly rare. In SLE, iridocyclitis may occur but is often mild. If uveitis is present, other causes should be considered.
Variants in more than 80 genetic loci associated with SLE have been identified. A complex balance of susceptibility and protective genes contributes to disease onset.
The overwhelming predominance in women suggests involvement of estrogen and other hormones. The high incidence during reproductive years supports this hypothesis.
Lupus retinopathy is associated with poor disease control. Positive anticardiolipin antibodies play an important role in the development of occlusive lesions 2).
Others: Anti-SS-A antibody, anti-SS-B antibody (evaluation of secondary Sjögren’s syndrome)
In particular, antinuclear antibodies are positive in most cases during the active phase and are useful for assessing disease activity. As a uveitis screening, the basic items recommended by the Uveitis Clinical Practice Guidelines (HLA-B27, chest X-ray, syphilis serology, QFT-3G, ACE, ANA) should also be checked 1).
Fundus examination: Evaluate for cotton-wool spots, hemorrhages, and vascular abnormalities under dilated pupils
Fluorescein fundus angiography (FFA): Important for evaluating retinal vasculitis and vascular occlusion. Can detect leakage, telangiectasia, occlusion, and microaneurysms
Indocyanine green (ICG) angiography: Detects choroidopathy not visualized by FFA as choroidal hyperfluorescence
OCT-A (Optical coherence tomography angiography): May be useful for monitoring retinal structural changes in subclinical retinopathy (research stage)
QHow to distinguish SLE retinopathy from diabetic retinopathy?
A
SLE retinopathy is more occlusive and tends to cause severe ischemia compared to diabetic retinopathy. History of diabetes or high HbA1c is important for differentiation. Fluorescein angiography shows intense leakage from retinal vessels and microvascular abnormalities in the acute phase of SLE.
Anticoagulation is necessary in cases with progressive vascular occlusion due to retinal vasculitis. Warfarin 2–5 mg/day is adjusted to achieve PT-INR 1.5–2.
Belimumab (Benlysta®): BLyS/BAFF inhibitor. Approved for SLE in Japan. The BLISS-52 trial confirmed a significant reduction in disease activity3)
Anifrolumab (Saphnelo®): Anti-IFNAR1 antibody (type I IFN receptor inhibitor). Approved in Japan in 2022. Significantly improves disease activity in moderate to severe SLE4)
Voclosporin (calcineurin inhibitor): Indicated for lupus nephritis
Retinal photocoagulation: If retinal neovascularization is confirmed by fluorescein angiography, perform immediately to prevent vitreous hemorrhage. Also performed prophylactically for extensive retinal vascular occlusion.
Serous retinal detachment: Identify leakage points from the retinal pigment epithelium by fluorescein angiography and perform retinal photocoagulation to those points.
For the first 5 years after starting treatment, only baseline examinations are needed. From year 5 onward, annual Humphrey 10-2 visual field testing, SD-OCT, and fundus autofluorescence are recommended. If high-risk factors are present, screening should begin before 5 years. If maculopathy is detected, the drug should be discontinued.
QHow often should regular examinations be performed in patients using hydroxychloroquine?
A
For the first 5 years after starting treatment, only baseline examinations are needed, but from year 5 onward, annual fundus examinations are recommended. Humphrey 10-2 visual field testing and SD-OCT are the primary screening methods. In patients with high-risk factors (reduced renal function, high dose, long-term use), screening should begin earlier5).
The pathogenesis of SLE is based on loss of self-tolerance and overproduction of autoantibodies.
Breakdown of self-tolerance: Genetic and environmental factors lead to loss of immune tolerance to self-antigens
T cell abnormalities: Helper T cells are overactivated, and regulatory immune cells are reduced
B cell maturation abnormalities: B cells mature more rapidly, and apoptosis is suppressed. Plasma cells become long-lived and produce excessive autoantibodies
Type I interferon pathway activation: B cell overactivation via BLyS/BAFF
Immune complex formation: Autoantibodies bind to nuclear, intranuclear, and cytoplasmic self-antigens, leading to release of inflammatory cytokines
Tissue damage: Chronic inflammation, immune complex deposition, and defective clearance of apoptotic cells cause tissue and organ damage
The pathological hallmark of SLE is vasculitis with fibrinoid necrosis of small blood vessels and capillaries. Fibrinoid material consists of fibrin, immune complexes, and complement.
The pathophysiology of lupus retinopathy involves a dual mechanism.
Immune complex vasculitis: Deposition of immune complexes on the vascular endothelium activates complement and releases inflammatory mediators, leading to non-perfusion and ischemia.
Thrombotic mechanism: Development of antiphospholipid antibody syndrome causes retinal vascular thrombosis. Positive anticardiolipin antibodies are associated with occlusive lupus retinopathy2).
In addition to chronic inflammation and immune complex deposition, the development of secondary Sjögren’s syndrome is the main cause. Autoimmune attack on the lacrimal gland reduces tear secretion.
QWhy is SLE retinopathy more likely to become severe than diabetic retinopathy?
A
SLE retinopathy involves a dual mechanism of immune complex vasculitis and antiphospholipid antibody-induced thrombosis, resulting in a more occlusive pathology. It tends to cause more severe ischemia than diabetic retinopathy, and occlusive vascular lesions are associated with worse visual prognosis.
7. Latest Research and Future Perspectives (Investigational Reports)
Optical coherence tomography angiography (OCT-A) is a new imaging method that can non-invasively evaluate subclinical retinal microvascular changes that are difficult to detect with conventional fluorescein angiography. It is expected to be applied to early detection and monitoring of SLE retinopathy, but its usefulness is still not fully studied.
In CD19-directed CAR-T therapy for refractory SLE, drug-free remission has been reported in a small number of treatment-resistant patients6). Long-term effects on ocular complications remain a future challenge.
Anifrolumab (type I IFN receptor inhibitor) improves disease control in active SLE and is expected to have indirect effects on ocular complications4). Individual efficacy data in the ophthalmology field are still being accumulated.
Stafford-Brady FJ, Urowitz MB, Gladman DD, Easterbrook M. Lupus retinopathy. Patterns, associations, and prognosis. Arthritis Rheum. 1988;31:1105-1110.
Navarra SV, Guzmán RM, Gallacher AE, et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: a randomised, placebo-controlled, phase 3 trial (BLISS-52). Lancet. 2011;377:721-731. doi:10.1016/s0140-6736(10)61354-2.
Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, Bae SC, Brohawn PZ, Pineda L, Berglind A, Tummala R, TULIP-2 Trial Investigators.. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med. 2020;382(3):211-221. doi:10.1056/nejmoa1912196. PMID:31851795.
Marmor MF, Kellner U, Lai TYY, et al. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy (2016 Revision). Ophthalmology. 2016;123:1386-1394.
Mackensen A, Müller F, Mougiakakos D, et al. Anti-CD19 CAR T cell therapy for refractory systemic lupus erythematosus. Nat Med. 2022;28:2124-2132. doi:10.1038/s41591-022-02017-5.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.