Scleritis is inflammation of deep blood vessels, such as the episcleral vascular plexus covering the superficial sclera and the intrascleral vascular plexus, accompanied by scleral edema and cellular infiltration. The sclera is a fibrous tissue with poor blood supply, and deep scleritis is a rare disease. It can be unilateral or bilateral, and causes are broadly classified into idiopathic, systemic disease-associated, infectious, and postoperative.
The incidence is reported to be 1.6–5.5 per 100,000 person-years 5). It is more common in women, with a peak age of onset in the 40s for non-necrotizing diffuse and nodular scleritis, and in the 60s for necrotizing scleritis. Bilateral involvement occurs in about 60% of necrotizing scleritis cases. Non-infectious scleritis accounts for the majority and is often seen as an ocular manifestation associated with systemic inflammatory diseases. Infectious scleritis is rare, accounting for 5–10% of all cases, but has a poor prognosis 7).
According to a multicenter survey based on the Japanese guidelines for uveitis clinical practice, among 3,810 patients visiting uveitis clinics, 235 (6.2%) had scleritis, making it the second most common condition after acute anterior uveitis (6.6%)8).
The classic Watson classification (Watson et al., 1976) is widely used for classifying scleritis based on clinical findings. It broadly divides scleritis into anterior and posterior types, with anterior scleritis further subdivided into three types based on morphology.
Classification
Disease type
Characteristics
Anterior scleritis
Diffuse
Most common. Diffuse hyperemia due to dilation and tortuosity of scleral vessels
Anterior scleritis
Nodular
Dark red scleral nodule. Commonly occurs at the limbal palpebral fissure area.
Anterior scleritis
Necrotizing (inflammatory)
Risk of scleral necrosis, thinning, and perforation
Diffuse scleritis is the most common, followed by nodular scleritis. Necrotizing scleritis and posterior scleritis are rare. Recurrences often present with the same disease type, but about 10% become more severe. If the underlying systemic disease is untreated, recurrences frequently occur at the same site of the sclera. About 10% of nodular scleritis cases progress to necrotizing scleritis over time.
A special type of necrotizing scleritis with almost no inflammatory symptoms is called scleromalacia perforans. It commonly occurs in patients with long-standing rheumatoid arthritis, and slow scleral thinning progresses without redness or pain. Although the English name includes “perforans” (perforating), in reality, the eyeball shape is often maintained by a thin fibrous membrane.
QWhat is the difference between episcleritis and scleritis?
A
Episcleritis is inflammation of the superficial vascular plexus, such as the Tenon’s capsule vascular plexus, with mild hyperemia, no pain, and no effect on vision. Scleritis is inflammation of the deep blood vessels, accompanied by severe eye pain and dark red hyperemia. The two can be differentiated by the fact that hyperemia in episcleritis subsides with 1:1000 epinephrine eye drops, while hyperemia in scleritis does not.
Smeller L, Toth-Molnar E, Sohar N. Optical Coherence Tomography: Focus on the Pathology of Macula in Scleritis Patients. J Clin Med. 2023;12(14):4825. Figure 1. PMID: 37510941; PMCID: PMC10381547; DOI: 10.3390/jcm12144825. License: CC BY 4.0.
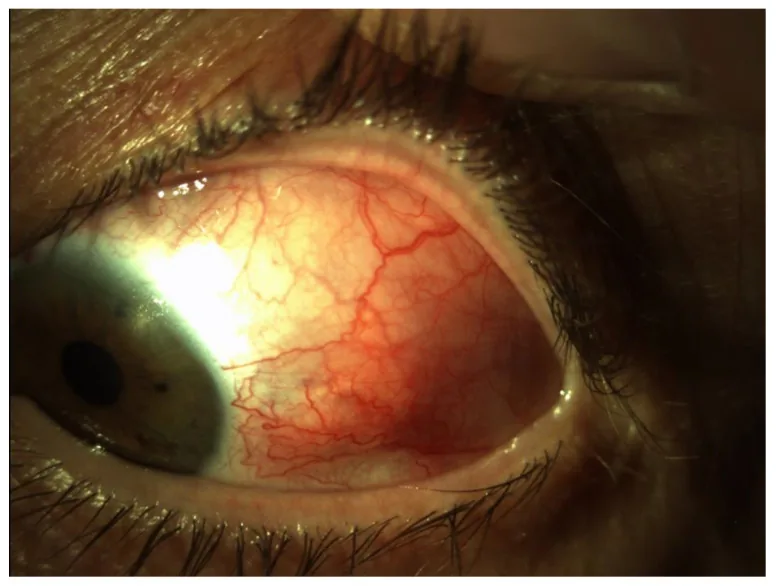
Anterior segment image showing diffuse hyperemia with marked dilation and tortuosity of deep vessels in the temporal sclera of the left eye. This corresponds to the patterns of hyperemia and vascular findings for each disease type discussed in the section “2. Main symptoms and clinical findings.”
Severe eye pain: Characterized by a boring deep pain. It may be severe enough to disturb sleep.
Radiating pain: Pain radiates to the ear, face, jaw, and temple. This is particularly prominent in diffuse scleritis.
Worsening at night and pain with eye movement: Pain worsens at night and with eye movement.
Tenderness: Patients often report tenderness upon palpation.
Hyperemia: The patient experiences hyperemia accompanied by throbbing severe pain.
Decreased visual acuity: In many cases, patients first become aware of symptoms when the condition has progressed to necrotizing scleritis or when the retina or optic nerve is affected in posterior scleritis.
Peculiarity of perforating scleromalacia: It follows a course in which a scleral necrosis focus suddenly appears in an eye that had almost no inflammatory symptoms, hyperemia, or pain, or the exposure of the uvea due to a scleral defect is noticed.
Clinical Findings (Findings Confirmed by the Physician During Examination)
Deep vascular dilation and tortuosity: Inflammation of the scleral vessels causes dilation of the episcleral and intrascleral vascular plexuses. The scleral vessels are immobile.
Purple-blue discoloration: This is a characteristic color change in scleritis. In contrast to the bright red of conjunctivitis or episcleritis, it appears dark red to purple-blue. It is easier to observe with the naked eye under natural light than with a slit lamp. In long-standing cases, localized or diffuse scleral thinning gives a bluish-black appearance.
Epinephrine eye drop test: Deep vascular congestion does not resolve with 1:1,000 epinephrine eye drops. Superficial conjunctival and episcleral congestion resolves, making this test useful for differentiating from conjunctivitis and episcleritis.
No palpebral conjunctival findings: Even in severe cases, there are no inflammatory findings in the palpebral conjunctiva, making it easy to differentiate from conjunctivitis.
Tenderness on palpation: This is an important aid in differentiating from conjunctivitis and episcleritis.
Differences in findings by disease type: Each disease type has distinct characteristics, which can generally be distinguished by slit-lamp examination at the initial visit.
Diffuse scleritis
Hyperemia: Diffuse, severe hyperemia due to dilation and tortuosity of scleral vessels. It may be localized to the entire circumference or one or more quadrants.
Pain: Severe pain radiating to the face or temple, often severe enough to disturb sleep.
Notable findings: No nodules, elevations, necrosis, or thinning. May be accompanied by conjunctival edema, eyelid swelling, anterior uveitis, and elevated intraocular pressure.
Nodular Scleritis
Nodules: Single or multiple dark red nodules. Commonly occur in the palpebral fissure near the limbus.
Palpation: The nodule is immobile and tender.
History: Many cases have a history of herpes zoster ophthalmicus. About 10% progress to necrotizing scleritis, but with early treatment, it heals leaving only small scars.
Necrotizing scleritis
Scleral necrosis: In the early stage, localized white to yellow avascular areas (scleral necrotic foci). Accompanied by marked dilation, tortuosity, and melting of scleral vessels.
Thinning: The uvea becomes thin enough to be visible, and if it progresses further, it can lead to perforation of the eyeball. The thinned area remains even after inflammation subsides.
Prognosis: The age of onset is in the 60s, and bilateral involvement accounts for about 60%. Without early treatment, blindness and difficulty in preserving the eyeball may occur.
Posterior scleritis
Epidemiology: The average age of onset is about 50 years, with a predilection for females, occurring approximately twice as often as in males. Bilateral involvement occurs in 30–40% of cases.
Fundus findings: Optic disc edema, choroidal folds, exudative retinal detachment, subretinal mass, and elevated intraocular pressure. Uveal effusion and secondary angle-closure glaucoma have also been reported.
Spreading symptoms: When complicated by external ophthalmoplegia, it causes diplopia, eye movement pain, proptosis, and ptosis.
When inflammation spreads from scleritis to the cornea, peripheral corneal infiltration and ulcers may occur. Anterior uveitis may also be present. Since scleritis almost invariably involves the episclera, findings of episcleritis are also mixed in.
Posterior scleritis is often diagnosed late because the lesion is located deep in the fundus. It may develop simultaneously from the anterior to posterior segments or with a time lag. About one-third of posterior scleritis cases are accompanied by anterior scleritis, and during the course of posterior scleritis, anterior scleritis is observed in approximately 70% of cases1). Cases with anterior scleritis are strongly associated with systemic diseases.
Optic disc edema: Occurs when inflammation spreads to the orbital tissues or optic nerve, requiring urgent treatment to prevent permanent vision loss.
B-mode ultrasound T-sign: Due to scleral thickening and sub-Tenon fluid accumulation, the boundary between the optic nerve and sclera appears angular on imaging1). This is the most specific ultrasound finding for posterior scleritis.
Misdiagnosis as choroidal tumor: Posterior scleritis may be referred to as a choroidal mass and is a cause of pseudomelanoma1).
Complication of extraocular myositis: When inflammation extends to the extraocular muscles, it causes diplopia, pain on eye movement, and redness around the insertion of the extraocular muscles.
QWhy is posterior scleritis often overlooked?
A
In posterior scleritis, anterior segment findings are scarce, and some patients present only with eye pain, headache, and decreased vision. Fundus findings such as choroidal folds and exudative retinal detachment are often definitively diagnosed only after evaluation of choroidal thickening using B-mode ultrasonography (T-sign) or OCT1). It may be mistaken for a choroidal tumor, so a comprehensive differential diagnosis is important.
Up to 50% of scleritis cases are associated with systemic autoimmune diseases. If the underlying systemic disease remains untreated, recurrence at the same site of the sclera is not uncommon. Necrotizing scleritis is often caused by rheumatic diseases, vasculitis, and hematologic disorders.
Collagen disease / rheumatic disease
Rheumatoid arthritis (RA): This is the most frequently associated systemic disease. It can cause necrotizing scleritis and scleromalacia perforans. It is typically seen in patients undergoing long-term treatment.
Other reported associations include sarcoidosis, Behçet’s disease, Crohn’s disease, ulcerative colitis, psoriatic arthritis, scleroderma, dermatomyositis, SAPHO syndrome, thyroid disease, aortitis syndrome, interstitial nephritis, Vogt-Koyanagi-Harada disease, and multiple sclerosis. Many cases of nodular scleritis have a history of herpes zoster ophthalmicus. Posterior scleritis may rarely present as an ocular manifestation of systemic lymphoma or multiple myeloma, requiring caution.
Infectious scleritis accounts for only 5–10% of all cases, but it has an extremely poor prognosis7). Approximately 50% of patients with infectious scleritis lose functional vision, and about 27% require enucleation or evisceration7).
Pseudomonas aeruginosa: The most common causative organism in Europe and the United States 7). Scleral necrosis progresses rapidly, presenting with purulent scleromalacia.
Nocardia species: Occurs after trauma or in immunocompromised patients2). Characterized by a chronic course with repeated remissions and relapses, and bacteria may remain deep even after nodules subside2).
Moraxella species: A rare causative bacterium, but it can develop as an opportunistic infection in immunocompromised states7).
Others: Infections such as fungi, tuberculosis, syphilis, and herpes virus have also been reported. In regions with a high prevalence of tuberculosis, it is recommended to rule out tuberculosis by tuberculin skin testing before systemic steroid administration.
Most cases of infectious scleritis are caused by exposed sutures or scleral buckle materials after ophthalmic surgery, and occur unilaterally.
Ophthalmic surgery can trigger necrotizing scleritis. Typical triggers include pterygium surgery, cataract surgery, scleral buckling, strabismus surgery, and trabeculectomy. It occurs particularly frequently after pterygium excision combined with mitomycin C. The onset period ranges from a few days to several years after surgery, and cases developing several years postoperatively are not rare.
The antimetabolites mitomycin C (MMC) and 5-fluorouracil (5-FU) have been used to prevent recurrence after pterygium surgery and to prevent scarring of glaucoma filtering blebs. MMC eye drops can cause scleral calcification or perforating scleral softening months to years after surgery, so their use as eye drops was discontinued in the 1980s. In current glaucoma and pterygium surgeries, a single intraoperative application of low-concentration MMC (0.02–0.04%) is standard, but postoperative pallor, vascular narrowing, and avascular zones may appear at the surgical site, potentially predisposing to future scleral softening.
QWhat systemic diseases are associated?
A
Rheumatoid arthritis is the most common, and other autoimmune diseases such as granulomatosis with polyangiitis (GPA), systemic lupus erythematosus, polyarteritis nodosa, relapsing polychondritis, Takayasu arteritis, and sarcoidosis are also associated. Up to 50% of patients with scleritis have some systemic disease. In necrotizing scleritis, the complication rate of rheumatic diseases, vasculitis, and hematologic diseases is even higher.
Naked-eye observation under natural light: In contrast to the bright red congestion of conjunctivitis or episcleritis, scleritis presents with a dark red to purplish-blue color. In long-standing cases, scleral thinning gives a bluish-black appearance. These color changes are easier to appreciate with naked-eye observation in a bright room than with a slit-lamp microscope.
Slit-lamp microscopy: Evaluates dilation and tortuosity of scleral vessels, presence of nodules, dark red appearance of scleral nodules, thinning, necrosis, and perforation. The absence of inflammatory findings in the palpebral conjunctiva is a distinguishing point from conjunctivitis.
Epinephrine instillation test: Deep scleral injection does not resolve with instillation of 1:1,000 epinephrine. This is important for differentiating from episcleritis and conjunctival injection.
Palpation: Touch the conjunctiva with a cotton swab or similar to check for tenderness. This helps differentiate conjunctivitis and episcleritis.
B-mode ultrasonography: Essential for diagnosing posterior scleritis. Characteristic findings include scleral thickening, scleral nodules, and the T-sign due to sub-Tenon fluid accumulation 1). It is also useful for differentiating from choroidal tumors.
Fluorescein scleral angiography: The presence or absence of scleral non-perfusion areas can help differentiate necrotizing scleritis.
CT/MRI: Used to evaluate scleral thickening in posterior scleritis and extraocular myositis, and to differentiate from intracranial lesions.
In necrotizing scleritis and refractory cases, screening for ANCA-associated vasculitis is particularly important3). In regions with a high prevalence of tuberculosis, tuberculin skin testing should be performed before systemic treatment in patients with scleritis resistant to topical steroid therapy.
Episcleritis: This is inflammation of the superficial blood vessels, with mild redness, no pain, and it resolves with phenylephrine eye drops.
Conjunctivitis: Hyperemia is most prominent in the conjunctival fornix and diminishes as it approaches the limbus. It is accompanied by discharge and abnormalities of the palpebral conjunctiva.
MALT lymphoma: A salmon-pink mass that commonly occurs in the conjunctival fornix. It is differentiated from scleritis by the fact that scleral vessels are not visible through the tumor because it lies beneath the conjunctiva.
Corneal diseases: It is necessary to differentiate between peripheral corneal infiltration spreading from scleritis and Mooren’s ulcer or staphylococcal peripheral corneal infiltration.
Tenon’s capsule inflammation: Considered a type of episcleritis, and differentiation between the two is difficult.
Orbital apex syndrome: In internal carotid artery-cavernous sinus fistula, congestion and dilation of conjunctival and scleral veins occur, accompanied by pulsating exophthalmos and diplopia.
Vogt-Koyanagi-Harada disease: This condition is difficult to differentiate from posterior scleritis. It presents with bilateral granulomatous anterior uveitis and choroidal thickening on OCT.
Choroidal tumor: Nodular lesions of posterior scleritis may be referred to as choroidal masses1), and differentiation is made by integrating ultrasound, MRI, and OCT.
Treatment for scleritis primarily involves steroids, with a stepwise combination of topical therapy, systemic therapy, immunosuppressive drugs, biologic agents, and surgical treatment depending on the type and severity of the disease. In cases with systemic complications, collaboration with rheumatology and collagen disease departments is essential.
Oral NSAIDs (first-line treatment)
Indications: Initial treatment for mild to moderate diffuse and nodular scleritis.
Prescription example: Celecoxib (COX-2 inhibitor) 100 mg twice daily orally, or indomethacin 50 mg three times daily. It is often highly effective for pain and also useful for controlling inflammation.
Caution: Watch for gastrointestinal bleeding, renal dysfunction, and asthma attacks. If there are no contraindications such as asthma, use it actively from the initial stage.
Topical Steroid Therapy
Eye drops: 0.1% betamethasone sodium phosphate eye drops are used 4 to 6 times daily. In some cases, betamethasone-fradiomycin ointment is also applied before bedtime.
Subconjunctival injection: Triamcinolone acetonide 40 mg/mL, 0.1 mL per dose (up to once a month), or dexamethasone 3.3 mg/mL, 0.3 mL per dose, every 1–2 weeks for several doses.
Caution: In necrotizing scleritis, inject while avoiding the thinned area.
Systemic steroid therapy
Oral administration: Prednisolone 0.5–1 mg/kg/day (for mild cases unresponsive to NSAIDs, a tapering regimen starting at 20–30 mg divided into two doses; for severe nodular, necrotizing, or posterior scleritis, a tapering regimen starting at 30–60 mg/day).
Pulse therapy: Methylprednisolone 1,000 mg/day is administered intravenously over 3 days, followed by a tapering regimen. This is indicated for necrotizing scleritis and severe cases.
Note: Tapering therapy is usually carried out over 1–2 weeks, and in severe cases, it may continue over a period of 2–3 months.
Immunosuppressants and Biologics
Cyclosporine: Start at 5 mg/kg/day in two divided doses, adjusting to maintain trough blood levels around 100–150 ng/mL. Monitor renal function with regular blood tests due to risk of kidney impairment.
Selection in the presence of systemic diseases: Methotrexate is often chosen for rheumatoid arthritis, and cyclophosphamide for systemic lupus erythematosus or systemic vasculitis. Azathioprine is less effective for scleritis.
Biologic agents: There are reports of administration of infliximab (anti-TNF-α antibody, Remicade®) and rituximab (anti-CD20 antibody, Rituxan®) in refractory cases3).
Treatment is initiated primarily with oral NSAIDs and 0.1% betamethasone eye drops. If the response is insufficient, immunosuppressive eye drops are added. If still inadequate, subconjunctival injection of 0.3 mL dexamethasone or 0.1–0.2 mL triamcinolone acetonide is performed, avoiding areas of scleral thinning. If local treatment is poorly effective, a tapering regimen of prednisolone 20–30 mg/day is continued for 1–2 weeks.
Severe nodular scleritis, necrotizing scleritis, and circumferential scleritis
Because scleral thinning and perforation can occur in a short period, initial treatment with oral prednisolone at 0.5–1 mg/kg/day is started. For cases that respond poorly to oral steroids or have repeated relapses, collaborate with a rheumatologist to select the optimal immunosuppressive drug for the associated systemic disease. When cyclosporine is used at 5 mg/kg/day in two divided doses, adjust the blood trough level to 100–150 ng/mL. Cyclosporine is contraindicated for scleritis associated with neuro-Behçet disease due to worsening of neurological symptoms. In severe cases, pulse therapy with intravenous methylprednisolone 1,000 mg/day for 3 days is performed after thoroughly ruling out infectious causes.
For scleritis resistant to immunosuppressive therapy, consider introducing biologic agents. TNF-α inhibitors have shown efficacy for sclerouveitis, but not all are effective for scleritis; etanercept has been reported to induce or exacerbate ocular inflammation including scleritis as a paradoxical reaction. Systemic examination before and after initiation and collaboration with internal medicine are essential when using immunosuppressive drugs and biologic agents.
Systemic administration of corticosteroids is the mainstay of treatment. Prednisolone is started at 30–50 mg/day and gradually tapered, divided into 2–3 doses for pain control. If anterior segment inflammation is present, topical corticosteroids are used concomitantly. If oral therapy fails to achieve inflammation control, steroid pulse therapy is considered after thoroughly ruling out infectious causes. If the disease is resistant to pulse therapy or relapses during tapering, active administration of immunosuppressive agents should be considered. Examples include azathioprine 1–2 mg/kg or methotrexate (Rheumatrex®) 6 mg/week. Necrotizing scleritis may require cyclophosphamide pulse therapy, and collaboration with an internist is particularly important.
Sub-Tenon triamcinolone acetonide injection (STTA) is also used for posterior scleritis, but it rarely carries a risk of causing circulatory disorders of the optic nerve and retinochoroid 6). In elderly patients with vascular fragility or in eyes with glaucoma, careful assessment of indications is necessary 6).
For infectious scleritis, the basic treatment is antimicrobial therapy based on identification of the causative organism and its susceptibility 2)7).
Selective treatment after identification of causative bacteria: For Nocardia infection, intensive amikacin eye drops and oral sulfamethoxazole-trimethoprim are used in combination for a long period. Surgical debridement may be repeated 2). For Pseudomonas aeruginosa infection, scleral necrosis (scleromalacia purulenta) progresses rapidly, so aggressive treatment with aminoglycoside or quinolone antibiotics as the mainstay and prompt surgical treatment should be performed.
Removal of exposed sutures and buckle materials: Exposed sutures that are the source of infection should be removed immediately. If scleral buckle materials do not respond to medical treatment, it is desirable to remove them within 1 to 2 weeks to prevent endophthalmitis.
Judgment on steroid use: Since steroid administration carries the risk of exacerbating infection, it should be used only after sufficiently ruling out infectious causes. If there is a response to antibiotics but inflammation persists, steroids may be used while monitoring white blood cell count and CRP trends.
Fungal infectious scleritis: Treatment follows the pharmacological treatment of fungal keratitis.
Necrotizing scleritis or scleromalacia perforans, and infectious scleritis unresponsive to medical treatment are indications for surgical treatment. If scleral necrosis or softening extends beyond a certain extent, it becomes difficult to restore the shape of the eyeball or maintain visual function, so early surgery is desirable while the necrotic area is still small.
The key points of surgery are the following three.
Complete resection of the scleral necrotic lesion including surrounding healthy tissue
Repair and reconstruction of the lesion site using preserved scleral graft: Preserved sclera is suitable as a graft material due to its strength and ability to maintain the shape of the eye wall. Preserved corneas often undergo melting.
Complete coverage of the transplanted scleral patch with conjunctiva
In cases involving extensive conjunctival necrosis or peripheral corneal ulcer, autologous conjunctival transplantation from the other eye or corneal epithelial transplantation is performed concurrently. Postoperative treatment includes oral cyclosporine and 1% Sandimmun® eye drops (hospital preparation) to promote graft survival and prevent recurrence of scleritis. Even in scleromalacia occurring after MMC or 5-FU use, given the current lack of confirmed efficacy of immunosuppressive eye drops or biologics, early preserved scleral grafting should be performed while the softened area is still small.
During long-term treatment with steroids and immunosuppressants, it is necessary to regularly monitor intraocular pressure, liver and kidney function, blood glucose levels, and blood cyclosporine concentration. If blood glucose levels rise, steroid treatment may need to be combined with insulin under systemic management in collaboration with an internal medicine specialist. For eye pain, analgesic and anti-inflammatory drugs are administered. Additionally, since scleritis can develop triggered by infection or infection-related allergy, initial treatment for first episodes also includes topical antibiotics and oral administration.
QHow is infectious scleritis treated?
A
Infectious scleritis has a poor prognosis, and early administration of antibiotics based on identification of the causative organism and its susceptibility is essential. In Pseudomonas aeruginosa infection, the disease progresses rapidly, so prompt initiation of potent antibiotic therapy and surgical debridement is necessary7). If exposed sutures or buckle materials are the cause, they should be removed promptly. Steroids should be used cautiously when infection is possible, as they carry a risk of exacerbating the infection.
The sclera is a fibrous tissue with poor vascularity, and the incidence of deep scleritis is low. However, because the sclera has innervation, once inflammation occurs, it causes severe eye pain. The sclera is thinnest at the attachment of the rectus muscles, approximately 0.3 mm, making it a common site for necrosis and perforation. Since the sclera lacks a barrier structure, drugs injected subconjunctivally or under the Tenon capsule can reach the intraocular space by diffusion, which is why subconjunctival steroid injection is a treatment option.
The pathology of scleritis associated with autoimmune diseases is characterized by zonal granulomatous necrosis. Fibrinoid material is seen at the center of the granuloma, surrounded by epithelioid cells and multinucleated giant cells.
In scleritis, infiltration of inflammatory cells including T cells and macrophages increases. T cells and macrophages infiltrate the deep episcleral tissue, and clusters of B cells form around blood vessels. Elevated expression of HLA-DR and IL-2 receptors on T cells suggests involvement of cell-mediated immune responses.
Plasma cells are involved in the production of matrix metalloproteinases (MMPs) and TNF-α. In necrotizing scleritis, vasculitis with fibrinoid necrosis is observed, along with neutrophil infiltration into the vessel walls. The pathogenesis of endogenous scleritis is suggested to involve immune mechanisms centered on cell-mediated immune responses.
Non-necrotizing scleritis (diffuse and nodular): Vasculitis is not prominent, and non-granulomatous inflammation is the main feature. In nodular type, fibrinoid necrosis is seen at the center of the lesion, surrounded by epithelioid cells arranged in a palisade.
Necrotizing scleritis: Small necrotic foci and non-granulomatous inflammation mainly composed of lymphocytes, plasma cells, and macrophages are observed. Vasculitis with fibrinoid necrosis and neutrophil infiltration are characteristic.
Infectious scleritis: Microabscesses form in addition to necrotizing inflammation. In Nocardia infection, even after the nodule subsides, bacteria remain deep within, leading to recurrent flare-ups 2).
Scleromalacia perforans: Occurs in patients with long-standing rheumatoid arthritis or related diseases. It presents as necrotic scleral plaques without congestion near the limbus, leading to gradual scleral thinning and exposure of the uvea.
A case series of 8 patients who developed posterior scleritis after COVID-19 vaccination or infection and were referred with a misdiagnosis of choroidal melanoma has been reported5). The mean interval from the last vaccination to onset was 132 days, and from COVID-19 infection to onset was 14 days5). Most cases resolved spontaneously within 2 months, with minimal impact on visual acuity5).
Rituximab for Refractory ANCA-Associated Scleritis
For ANCA-associated necrotizing scleritis resistant to conventional immunosuppressive therapy (steroids + cyclophosphamide), cases have been reported in which rituximab (anti-CD20 antibody) was effective for both remission induction and maintenance 3). Long-term follow-up studies demonstrating the efficacy of rituximab for ocular lesions in the granulomatosis with polyangiitis type of ANCA-associated vasculitis are also accumulating 3).
There have been reports of scleritis as an ocular manifestation of Takayasu arteritis, and it requires attention as a diagnostic clue for systemic aortic inflammatory syndrome 4). In young women with necrotizing scleritis, it is important to rule out Takayasu arteritis.
QIs there a link with COVID-19?
A
A case series of posterior scleritis developing after COVID-19 vaccination or infection has been reported5). However, a causal relationship has not been proven, and most cases follow a course of spontaneous resolution. COVID-19-related posterior scleritis may be mistaken for a choroidal tumor, and awareness of it as a differential diagnosis is important5).
Babu N, Kumar K, Upadhayay A, Kohli P. Nodular posterior scleritis - The great masquerader. Taiwan journal of ophthalmology. 2021;11(4):408-412. doi:10.4103/tjo.tjo_20_21. PMID:35070674; PMCID:PMC8757530.
Chauhan K, Murthy SI, Mitra S. Demystifying nocardial scleritis. BMJ case reports. 2023;16(11). doi:10.1136/bcr-2023-255730. PMID:38011958; PMCID:PMC10685915.
Tahavvori M, Fekri S, Hassanpour K, Sadoughi MM, Javadi M. Isolated ANCA-associated scleritis successfully treated with systemic rituximab; a case report and review of literature. BMC ophthalmology. 2025;25(1):176. doi:10.1186/s12886-025-04027-6. PMID:40197146; PMCID:PMC11974155.
Chittipolu S, Kennard JL, Tumma RS, Doyle AR. Scleritis in Takayasu Arteritis. Cureus. 2023;15(4):e37724. doi:10.7759/cureus.37724. PMID:37206528; PMCID:PMC10191460.
Negretti GS, Zeiger JS, Cherkas E, Shields CL. Posterior scleritis following COVID-19 vaccination or infection simulating uveal melanoma in 8 consecutive patients. Eye (London, England). 2024;38(1):185-191. doi:10.1038/s41433-023-02656-z. PMID:37422535; PMCID:PMC10764359.
Akada M, Muraoka Y, Morooka S, Ishihara K, Hata M, Tsujikawa A. SEVERE CIRCULATORY DISTURBANCE IN OPTIC DISK, RETINA, AND CHOROID AFTER SUB-TENON TRIAMCINOLONE ACETONIDE INJECTION FOR POSTERIOR SCLERITIS. Retinal cases & brief reports. 2025;19(6):789-792. doi:10.1097/ICB.0000000000001642. PMID:39058980; PMCID:PMC12570599.
Dallinga M, Murtagh P, Powell S, Murphy CC. Moraxella nonliquefaciens-associated infectious scleritis. BMJ case reports. 2023;16(5). doi:10.1136/bcr-2022-254113. PMID:37221000; PMCID:PMC10230883.