La sclerite è un’infiammazione dei vasi profondi, inclusi i plessi vascolari episclerale e intrasclerale, con edema e infiltrazione cellulare della sclera. La sclera è un tessuto fibroso scarsamente vascolarizzato e la sclerite profonda è una malattia rara. Può essere unilaterale o bilaterale, e le cause possono essere idiopatiche, associate a malattie sistemiche, infettive o post-operatorie.
L’incidenza è riportata tra 1,6 e 5,5 per 100.000 persone-anno 5). È più comune nelle donne. Per la sclerite diffusa e nodulare non necrotizzante, l’età tipica è intorno ai 40 anni; per la sclerite necrotizzante, intorno ai 60 anni. Il tasso di coinvolgimento bilaterale nella sclerite necrotizzante è di circa il 60%. La maggior parte dei casi è non infettiva, spesso associata a malattie infiammatorie sistemiche. La sclerite infettiva è rara (5-10%) ma con prognosi infausta 7).
Secondo un’indagine multicentrica giapponese basata sulle linee guida per l’uveite, su 3.810 pazienti visitati in ambulatorio per uveite, 235 (6,2%) presentavano sclerite, rendendola la seconda causa più frequente dopo l’uveite anteriore acuta (6,6%) 8).
Per la classificazione clinica della sclerite, è ampiamente utilizzata la classificazione classica di Watson (Watson et al., 1976). Essa distingue la sclerite anteriore e posteriore, e suddivide la sclerite anteriore in tre tipi in base alla morfologia.
Classificazione
Tipo
Caratteristiche
Sclerite anteriore
Diffusa
La più frequente. Iperemia diffusa dovuta a dilatazione e tortuosità dei vasi sclerali.
Sclerite anteriore
Nodulare
Nodulo sclerale rosso scuro. Frequente a livello del limbo e della rima palpebrale
Sclerite anteriore
Necrotizzante (infiammatoria)
Rischio di necrosi sclerale, assottigliamento, perforazione
La sclerite diffusa è la più frequente, seguita dalla sclerite nodulare. La sclerite necrotizzante e la sclerite posteriore sono rare. In caso di recidiva, spesso si manifesta lo stesso tipo di malattia, ma circa il 10% dei casi peggiora. Se la malattia sistemica sottostante non viene trattata, le recidive si verificano frequentemente nello stesso sito della sclera. Circa il 10% delle scleriti nodulari evolve in sclerite necrotizzante nel corso del tempo.
Un tipo speciale di sclerite necrotizzante con quasi nessun sintomo infiammatorio è chiamato scleromalacia perforante (scleromalacia perforans). Si verifica frequentemente in pazienti con artrite reumatoide di lunga durata e un lento assottigliamento sclerale progredisce senza arrossamento o dolore. Sebbene il nome inglese contenga “perforans” (perforazione), la forma del bulbo oculare è spesso mantenuta da una sottile membrana fibrosa.
QQual è la differenza tra episclerite e sclerite?
A
L’episclerite è un’infiammazione dei plessi vascolari superficiali, come il plesso della capsula di Tenone. L’arrossamento è lieve, indolore e non influisce sulla vista. La sclerite è un’infiammazione dei vasi profondi, accompagnata da forte dolore oculare e arrossamento rosso scuro. L’instillazione di epinefrina diluita 1:1000 fa regredire l’arrossamento dell’episclerite, ma non quello della sclerite, consentendo la diagnosi differenziale.
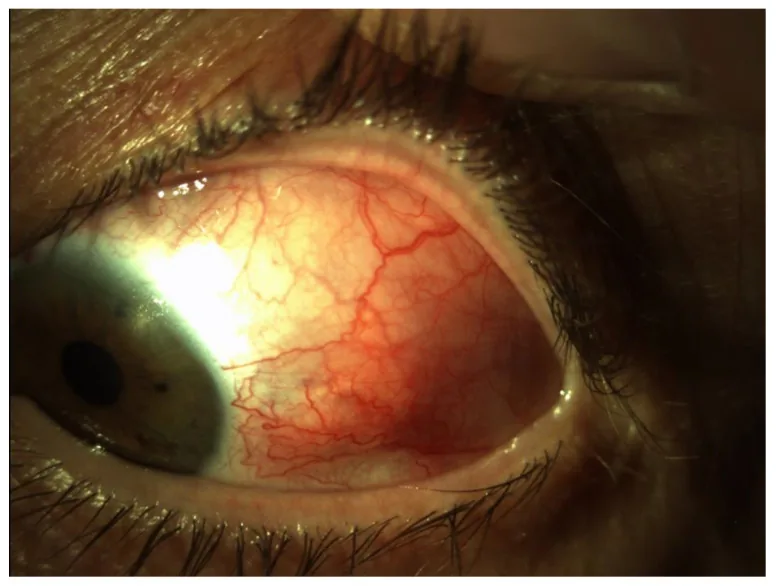
Smeller L, Toth-Molnar E, Sohar N. Optical Coherence Tomography: Focus on the Pathology of Macula in Scleritis Patients. J Clin Med. 2023;12(14):4825. Figure 1. PMID: 37510941; PMCID: PMC10381547; DOI: 10.3390/jcm12144825. License: CC BY 4.0.
Immagine del segmento anteriore dell’occhio sinistro che mostra arrossamento diffuso con marcata dilatazione e tortuosità dei vasi profondi della sclera temporale. Corrisponde ai pattern di arrossamento e ai reperti vascolari delle diverse forme di sclerite trattate nella sezione «2. Principali sintomi e segni clinici».
Forte dolore oculare: Caratteristico è un dolore profondo e trafittivo (boring). Il dolore può essere così intenso da disturbare il sonno.
Dolore irradiato: Il dolore si irradia all’orecchio, al viso, alla mandibola e alla tempia. È particolarmente evidente nella sclerite diffusa.
Peggioramento notturno e dolore ai movimenti oculari: Il dolore peggiora di notte e con i movimenti oculari.
Dolore alla palpazione: Spesso i pazienti riferiscono dolore alla palpazione.
Arrossamento: Il paziente avverte arrossamento accompagnato da un forte dolore pulsante.
Riduzione della vista: Spesso viene notata solo nei casi gravi che progrediscono verso la sclerite necrotizzante, o nella sclerite posteriore quando la retina o il nervo ottico sono danneggiati.
Particolarità della scleromalacia perforante: Si manifesta con la comparsa improvvisa di un focolaio di necrosi sclerale in un occhio senza arrossamento o dolore, oppure con la scoperta di un’esposizione uveale dovuta a un difetto sclerale.
Segni clinici (reperti riscontrati dal medico all’esame)
Dilatazione e tortuosità dei vasi profondi: L’infiammazione dei vasi sclerali provoca la dilatazione dei plessi vascolari episclerali e intrasclerali. I vasi sclerali sono immobili.
Colorito violaceo-bluastro: È un cambiamento di colore caratteristico della sclerite. A differenza del rosso vivo della congiuntivite o dell’episclerite, la sclerite presenta un colore rosso scuro o violaceo-bluastro. È più facilmente osservabile a occhio nudo sotto luce naturale che al microscopio a lampada a fessura. Nei casi di lunga durata, l’assottigliamento sclerale localizzato o diffuso conferisce un aspetto bluastro-nerastro.
Test con epinefrina collirio: L’instillazione di epinefrina 1:1000 non fa regredire l’iperemia dei vasi profondi. L’iperemia congiuntivale ed episclerale superficiale regredisce, utile per differenziare la sclerite dalla congiuntivite o dall’episclerite.
Assenza di segni congiuntivali palpebrali: Anche nei casi gravi non si riscontrano segni infiammatori della congiuntiva palpebrale, facilitando la distinzione dalla congiuntivite.
Dolore alla palpazione: Importante ausilio per differenziare la sclerite dalla congiuntivite o dall’episclerite.
Differenze in base al tipo: Ogni tipo presenta caratteristiche distinte, generalmente identificabili all’esame con lampada a fessura alla prima visita.
Sclerite diffusa
Iperemia: Rossore intenso diffuso per dilatazione e tortuosità dei vasi sclerali. Può essere circolare o localizzato a uno o più quadranti.
Dolore: Dolore intenso che si irradia al viso o alla tempia, tale da disturbare il sonno.
Reperti particolari: Assenza di noduli, rilievi, necrosi o assottigliamento. Possono associarsi edema congiuntivale, gonfiore palpebrale, uveite anteriore o ipertensione oculare.
Sclerite nodulare
Noduli: Noduli rosso scuro singoli o multipli, frequenti vicino al limbo nella regione della rima palpebrale.
Palpazione: I noduli sono fissi e dolenti alla palpazione.
Anamnesi: Spesso precedente herpes zoster oftalmico. Circa il 10% evolve in sclerite necrotizzante, ma con trattamento precoce guarisce lasciando solo piccole cicatrici.
Sclerite necrotizzante
Necrosi sclerale: Inizialmente aree avascolari bianco-giallastre localizzate (focolai di necrosi sclerale). Marcata dilatazione e tortuosità dei vasi sclerali con fusione.
Assottigliamento: Assottigliamento fino a rendere visibile l’uvea, con possibile perforazione oculare in fase avanzata. L’assottigliamento persiste anche dopo la risoluzione dell’infiammazione.
Prognosi: L’età di insorgenza è avanzata, intorno ai 60 anni, e circa il 60% dei casi è bilaterale. Senza un trattamento precoce, si verifica cecità e diventa difficile preservare il bulbo oculare.
Sclerite posteriore
Epidemiologia: L’età media di insorgenza è di circa 50 anni, con una prevalenza nelle donne (circa il doppio rispetto agli uomini). La bilateralità è del 30-40%.
Reperti del fondo oculare: Edema del disco ottico, pieghe coroidali, distacco di retina essudativo, massa sottoretinica, ipertensione oculare. Sono stati riportati anche versamento uveale e glaucoma secondario ad angolo chiuso.
Sintomi di diffusione: In caso di miosite dei muscoli extraoculari associata, si manifestano diplopia, dolore ai movimenti oculari, esoftalmo e ptosi palpebrale.
Quando l’infiammazione si diffonde dalla sclera alla cornea, si verificano infiltrati limbari e ulcere. Può anche essere associata un’uveite anteriore. La sclerite coinvolge quasi sempre anche l’episclera, quindi sono presenti anche segni di episclerite.
La sclerite posteriore viene generalmente diagnosticata tardivamente perché le lesioni sono profonde nel fondo oculare. Può insorgere contemporaneamente alla sclerite anteriore o in tempi diversi. Circa un terzo delle scleriti posteriori è associato a sclerite anteriore e nel corso della sclerite posteriore si osserva sclerite anteriore in circa il 70% dei casi1). I casi con sclerite anteriore sono più fortemente associati a malattie sistemiche.
Distacco di retina essudativo: Distacco sieroso della retina al polo posteriore1).
Edema del disco ottico: Compare quando l’infiammazione si diffonde ai tessuti orbitari o al nervo ottico, richiedendo un trattamento urgente per evitare una perdita visiva permanente.
Segno T all’ecografia in modalità B: A causa dell’ispessimento sclerale e dell’accumulo di liquido sotto la capsula di Tenone, il confine tra nervo ottico e sclera appare angolato1). È il segno ecografico più specifico della sclerite posteriore.
Confusione con tumore coroidale: La sclerite posteriore può essere inviata come massa coroidale ed è una causa di pseudomelanoma1).
QPerché la sclerite posteriore viene spesso trascurata?
A
Nella sclerite posteriore, i segni del segmento anteriore sono scarsi; alcuni pazienti si presentano solo con dolore oculare, cefalea o riduzione della vista. Le pieghe coroidali e il distacco essudativo della retina come reperti del fondo oculare vengono spesso diagnosticati con certezza solo dopo la valutazione del segno T all’ecografia in modalità B e dello spessore coroidale mediante OCT1). Può anche essere scambiato per un tumore coroidale, quindi è importante una diagnosi differenziale completa.
Fino al 50% delle scleriti sono associate a una malattia autoimmune sistemica. Se la malattia sistemica sottostante non viene trattata, non sono rare le recidive ripetute nello stesso sito sclerale. Le cause della sclerite necrotizzante sono spesso malattie reumatiche, vasculiti e malattie del sangue.
Malattie del collageno e reumatiche
Artrite reumatoide (AR) : È la malattia sistemica più frequentemente associata. Può causare sclerite necrotizzante o scleromalacia perforante. Tipica nei pazienti in terapia a lungo termine.
Altre malattie associate includono: sarcoidosi, malattia di Behçet, morbo di Crohn, colite ulcerosa, artrite psoriasica, sclerodermia, dermatomiosite, sindrome SAPHO, malattie tiroidee, sindrome dell’arco aortico, nefrite interstiziale, malattia di Vogt-Koyanagi-Harada, sclerosi multipla, ecc. Nella sclerite nodulare, è frequente una storia di herpes zoster oftalmico. Nella sclerite posteriore, bisogna prestare attenzione perché raramente può essere una manifestazione oculare di linfoma sistemico o mieloma multiplo.
La sclerite infettiva rappresenta solo il 5-10% di tutti i casi, ma ha una prognosi estremamente sfavorevole 7). Circa il 50% dei pazienti con sclerite infettiva perde la vista funzionale e circa il 27% va incontro a enucleazione o eviscerazione7).
Pseudomonas aeruginosa (Pseudomonas aeruginosa) : L’agente patogeno più comune in Europa e Nord America 7). La necrosi sclerale progredisce rapidamente, assumendo l’aspetto di una scleromalacia purulenta.
Genere Nocardia : Si manifesta dopo un trauma o in pazienti immunocompromessi 2). È caratteristico un decorso cronico con remissioni e riacutizzazioni; anche dopo la risoluzione dei noduli, i batteri persistono in profondità 2).
Genere Moraxella : Agente patogeno raro, ma si manifesta come infezione opportunistica in stati di immunodeficienza 7).
Altri : Sono stati riportati anche casi di infezione da funghi, tubercolosi, sifilide, virus herpes, ecc. Nelle regioni ad alta incidenza di tubercolosi, si raccomanda di escludere la tubercolosi mediante test tubercolinico prima della somministrazione sistemica di steroidi.
La maggior parte delle scleriti infettive è causata da suture esposte o materiale di cerchiaggio sclerale dopo chirurgia oculare e si manifesta in modo unilaterale.
Sclerite necrotizzante indotta da chirurgia (SINS)
Una sclerite necrotizzante può verificarsi in seguito a chirurgia oculare. Gli interventi tipici sono la chirurgia dello pterigio, la chirurgia della cataratta, il cerchiaggio sclerale, la chirurgia dello strabismo e la trabeculectomia. È particolarmente frequente dopo l’escissione dello pterigio con mitomicina C. Il tempo di insorgenza varia da pochi giorni a diversi anni dopo l’intervento; non sono rari i casi che si manifestano anni dopo.
Gli antimetaboliti mitomicina C (MMC) e 5-fluorouracile (5-FU) sono stati utilizzati per prevenire la recidiva dello pterigio e la cicatrizzazione delle bolle filtranti nel glaucoma. L’uso di colliri a base di MMC è stato sospeso negli anni ‘80 perché poteva causare calcificazione sclerale o scleromalacia perforante mesi o anni dopo l’intervento. Nella chirurgia attuale del glaucoma e dello pterigio, si utilizza principalmente una singola applicazione intraoperatoria di MMC a bassa concentrazione (0,02-0,04%) per breve tempo. Tuttavia, dopo l’intervento possono comparire pallore sclerale, restringimento vascolare e zone avascolari nel sito chirurgico, che possono costituire un terreno fertile per una futura scleromalacia.
QQuali malattie sistemiche sono associate?
A
L’artrite reumatoide è la più frequente, seguita da granulomatosi con poliangioite (GPA), lupus eritematoso sistemico, poliarterite nodosa, policondrite ricorrente, arterite di Takayasu e sarcoidosi. Fino al 50% dei pazienti con sclerite presenta una malattia sistemica. Nella sclerite necrotizzante, la frequenza di malattie reumatiche, vasculiti e malattie del sangue è ancora più elevata.
Osservazione a occhio nudo sotto luce naturale: A differenza dell’arrossamento rosso vivo della congiuntivite o dell’episclerite, la sclerite presenta un colore rosso scuro o violaceo-bluastro. Nei casi di lunga durata, l’assottigliamento sclerale conferisce un aspetto blu-nero. Questi cambiamenti di colore sono più facilmente apprezzabili a occhio nudo in ambiente luminoso che al microscopio a lampada a fessura.
Esame con lampada a fessura: Valuta la dilatazione e la tortuosità dei vasi sclerali, la presenza di noduli, l’aspetto rosso scuro dei noduli sclerali, l’assottigliamento, la necrosi e la perforazione. L’assenza di segni infiammatori sulla congiuntiva palpebrale è un punto di differenziazione dalla congiuntivite.
Test con epinefrina: L’instillazione di epinefrina diluita 1:1000 non fa regredire l’iperemia sclerale profonda. Importante per la differenziazione dall’episclerite e dall’iperemia congiuntivale.
Palpazione: Verificare la presenza di dolorabilità alla pressione toccando la congiuntiva con un bastoncino cotonato. Aiuta nella differenziazione da congiuntivite ed episclerite.
Ecografia in modalità B: Indispensabile per la diagnosi di sclerite posteriore. L’ispessimento sclerale, i noduli sclerali e il segno a T dovuto all’accumulo di liquido sotto la capsula di Tenone sono caratteristici 1). Utile anche per differenziare i tumori coroideali.
Angiografia sclerale con fluoresceina: Consente di differenziare la sclerite necrotizzante in base alla presenza di aree di non perfusione sclerale.
TC e RM: Utilizzate per valutare l’ispessimento sclerale nella sclerite posteriore, la miosite dei muscoli extraoculari e per differenziare le lesioni intracraniche.
Infezioni: Sierologia per sifilide, test Quantiferon, test tubercolinico
Altro: Enzima di conversione dell’angiotensina (ACE), lisozima, acido urico sierico
Nella sclerite necrotizzante e nei casi refrattari, la ricerca di una vasculite associata ad ANCA è particolarmente importante 3). Nelle regioni ad alta prevalenza di tubercolosi, in caso di sclerite resistente al trattamento steroideo locale, deve essere eseguito un test tubercolinico prima della somministrazione sistemica.
Episclerite: infiammazione dei vasi superficiali, iperemia lieve, assenza di dolore, regressione dopo instillazione di fenilefrina.
Congiuntivite: l’iperemia è più marcata nel fornice congiuntivale e diminuisce avvicinandosi al limbo. È accompagnata da secrezione oculare e anomalie della congiuntiva palpebrale.
Linfoma MALT: massa di colore rosa salmone che si verifica frequentemente nel fornice congiuntivale. Si differenzia dalla sclerite perché i vasi sclerali non sono visibili a causa della localizzazione sottocongiuntivale della massa.
Malattie corneali: è necessario differenziare l’infiltrato corneale periferico derivante dalla sclerite dall’ulcera di Mooren o dall’infiltrato corneale periferico stafilococcico.
Tenonite: considerata un tipo di episclerite, la distinzione tra le due è difficile.
Sindrome dell’apice orbitario: nella fistola carotido-cavernosa si verifica congestione e dilatazione delle vene congiuntivali e sclerali, accompagnate da esoftalmo pulsante e diplopia.
Malattia di Vogt-Koyanagi-Harada: difficile da differenziare dalla sclerite posteriore. Si presenta bilateralmente con uveite anteriore granulomatosa e ispessimento coroidale all’OCT.
Tumore coroidale: le lesioni nodulari della sclerite posteriore possono essere riferite come masse coroidali 1); la differenziazione viene effettuata mediante ecografia, RM e OCT.
Il trattamento della sclerite si basa principalmente sugli steroidi, con una combinazione graduale di terapia locale, sistemica, immunosoppressori, farmaci biologici e trattamento chirurgico in base al tipo e alla gravità. In caso di malattia sistemica associata, è indispensabile la collaborazione con reumatologi e specialisti in malattie del collageno.
FANS orali (prima linea)
Indicazioni: trattamento iniziale della sclerite diffusa o nodulare lieve-moderata.
Esempio di prescrizione: celecoxib (inibitore della COX-2) 100 mg due volte al giorno, o indometacina 50 mg tre volte al giorno. Spesso molto efficace per il dolore e anche efficace per il controllo dell’infiammazione.
Attenzione : prestare attenzione a sanguinamento gastrointestinale, compromissione renale e attacchi d’asma. In assenza di controindicazioni come l’asma, associare attivamente fin dall’inizio.
Terapia steroidea locale
Collirio : utilizzare betametasone sodio fosfato 0,1% collirio 4-6 volte al giorno. A seconda del caso, associare un unguento a base di betametasone e fradiomicina prima di coricarsi.
Iniezione sottocongiuntivale : triamcinolone acetonide 40 mg/mL, 0,1 mL per iniezione (fino a una volta al mese), o desametasone 3,3 mg/mL, 0,3 mL per iniezione, ogni 1-2 settimane per alcune volte.
Attenzione : nella sclerite necrotizzante, iniettare evitando le aree assottigliate.
Terapia steroidea sistemica
Orale : prednisolone 0,5-1 mg/kg/die (nei casi lievi con fallimento dei FANS, 20-30 mg in due dosi con riduzione graduale; nella sclerite nodulare grave, necrotizzante o posteriore, 30-60 mg/die con riduzione graduale).
Terapia pulsata : metilprednisolone 1.000 mg/die per via endovenosa per 3 giorni, seguita da riduzione graduale. Indicata nella sclerite necrotizzante e nei casi gravi.
Attenzione : la riduzione graduale viene solitamente effettuata in 1-2 settimane o più, nei casi gravi prosegue per 2-3 mesi.
Immunosoppressori e farmaci biologici
Ciclosporina : iniziare con 5 mg/kg/die per via orale in due dosi, regolare per mantenere un livello trough ematico intorno a 100-150 ng/mL. Prestare attenzione alla compromissione renale e richiedere esami del sangue regolari.
Scelta in caso di malattie sistemiche associate : per l’artrite reumatoide si sceglie spesso il metotrexato, per il lupus eritematoso sistemico o la vasculite sistemica si preferisce la ciclofosfamide. L’azatioprina è meno efficace nella sclerite.
Farmaci biologici : esistono segnalazioni di utilizzo di infliximab (anticorpo anti-TNF-α, Remicade®) e rituximab (anticorpo anti-CD20, Rituxan®) in casi refrattari3).
Protocollo terapeutico in base al tipo di sclerite
Il trattamento inizia con FANS orali e collirio a base di betametasone 0,1%. Se la risposta è insufficiente, si aggiungono colliri immunosoppressori; se ancora insufficiente, si esegue un’iniezione sottocongiuntivale di 0,3 mL di desametasone o 0,1-0,2 mL di triamcinolone acetonide, evitando le aree scleriche assottigliate. In caso di scarsa risposta al trattamento locale, si prosegue con una terapia decrescente di prednisolone 20-30 mg/die per 1-2 settimane.
A causa del rischio di assottigliamento e perforazione sclerale a breve termine, si inizia fin dall’inizio con prednisolone orale 0,5-1 mg/kg/die. Nei casi con risposta insufficiente agli steroidi orali o con recidive frequenti, in collaborazione con un reumatologo si sceglie l’immunosoppressore più adatto alla malattia sistemica associata. In caso di uso concomitante di ciclosporina 5 mg/kg/die in due dosi orali, si regola il livello ematico trough tra 100 e 150 ng/mL. Per la sclerite associata alla malattia di Behçet neurologica, la ciclosporina è controindicata perché aggrava i sintomi neurologici. Nei casi gravi, dopo aver escluso adeguatamente una causa infettiva, si esegue una terapia pulsata con metilprednisolone 1.000 mg/die per via endovenosa per 3 giorni.
Per la sclerite resistente al trattamento immunosoppressivo, si considera l’introduzione di farmaci biologici. Gli inibitori del TNF-α hanno mostrato efficacia nella sclero-uveite, ma non tutti sono efficaci per la sclerite; con etanercept sono stati riportati casi di reazione paradossa con induzione o peggioramento dell’infiammazione oculare, inclusa la sclerite. L’uso di immunosoppressori e biologici richiede esami sistemici prima e dopo l’introduzione e la collaborazione con un internista.
La somministrazione sistemica di corticosteroidi è il cardine del trattamento. Si inizia con prednisolone 30-50 mg/die in 2-3 dosi frazionate per il controllo del dolore, poi si riduce gradualmente. In caso di infiammazione del segmento anteriore, si associano colliri corticosteroidei. Se l’infiammazione non si attenua con la terapia orale, dopo aver escluso una causa infettiva si esegue una terapia pulsata con steroidi. In caso di resistenza alla terapia pulsata o di recidiva durante la riduzione, si considera la somministrazione attiva di immunosoppressori. Esempi di prescrizione includono azatioprina 1-2 mg/kg, metotrexato (Rheumatrex®) 6 mg/settimana. Nella sclerite necrotizzante può essere necessaria una terapia pulsata con ciclofosfamide, e la collaborazione con un internista è particolarmente importante.
L’iniezione sottotenoniana di triamcinolone acetonide (STTA) viene utilizzata anche per la sclerite posteriore, ma comporta un raro rischio di disturbi circolatori del nervo ottico e della corioretina6). Negli anziani con fragilità vascolare o nei pazienti con glaucoma è necessaria un’attenta valutazione dell’indicazione6).
Nella sclerite infettiva, il trattamento di base è la terapia antibiotica basata sull’identificazione dell’agente patogeno e sulla sua sensibilità2)7).
Trattamento selettivo dopo identificazione del patogeno: Nell’infezione da Nocardia, si associano colliri di amikacina potenziati e sulfametossazolo-trimetoprim per via orale per un lungo periodo. Può essere necessario un debridement chirurgico ripetuto2). Nell’infezione da Pseudomonas aeruginosa, la necrosi sclerale (scleromalacia purulenta) progredisce rapidamente, pertanto è necessario un trattamento intensivo con aminoglicosidi o fluorochinoloni e un intervento chirurgico tempestivo.
Rimozione di suture esposte e materiale del cerchiaggio: Le suture esposte che sono fonte di infezione devono essere rimosse immediatamente. Anche il materiale del cerchiaggio sclerale, se non risponde alla terapia farmacologica, è opportuno rimuoverlo entro 1-2 settimane per prevenire l’endoftalmite.
Decisione sull’uso concomitante di steroidi: Poiché la somministrazione di steroidi comporta il rischio di aggravare l’infezione, devono essere utilizzati solo dopo aver escluso adeguatamente una causa infettiva. Se il paziente risponde agli antibiotici ma l’infiammazione persiste, gli steroidi possono essere utilizzati monitorando la conta leucocitaria e la PCR.
Sclerite infettiva fungina: Il trattamento segue la terapia farmacologica della cheratite fungina.
La sclerite necrotizzante o la scleromalacia perforante, e la sclerite infettiva che non risponde alla terapia medica sono indicazioni al trattamento chirurgico. Quando la necrosi o l’area di rammollimento della sclera supera una certa estensione, diventa difficile ripristinare la forma del bulbo oculare e mantenere la funzione visiva. Pertanto, è auspicabile un intervento chirurgico precoce finché l’area necrotica è piccola.
I punti chiave dell’intervento chirurgico sono i seguenti tre:
Resezione completa del focolaio di necrosi sclerale, compreso il tessuto sano circostante
Riparazione e riempimento della lesione mediante trapianto di sclera conservata: La sclera conservata è un materiale di riempimento adatto per la sua resistenza e il mantenimento della morfologia della parete oculare. La cornea conservata spesso si lisa.
Copertura completa del lembo sclerale trapiantato con la congiuntiva
In caso di necrosi congiuntivale estesa o ulcera corneale periferica, si associa un autotrapianto congiuntivale dall’altro occhio o un trapianto epiteliale corneale. Nel trattamento postoperatorio si utilizzano ciclosporina per via orale e colliri di Sandimmun® all’1% (preparazione ospedaliera) per favorire l’attecchimento del trapianto e prevenire la recidiva della sclerite. Anche nella scleromalacia insorta dopo l’uso di MMC o 5-FU, in assenza di prove certe di efficacia di colliri immunosoppressori o agenti biologici, si dovrebbe eseguire un precoce trapianto di sclera conservata finché l’area di rammollimento è piccola.
Gestione sistemica e monitoraggio degli effetti collaterali
Nel trattamento a lungo termine con steroidi e immunosoppressori, è necessario monitorare regolarmente la pressione intraoculare, la funzionalità epatica e renale, la glicemia e la concentrazione ematica di ciclosporina. In caso di iperglicemia, può essere necessaria una terapia steroidea con insulina sotto gestione sistemica in collaborazione con uno specialista in medicina interna. Per il dolore oculare si somministrano analgesici e antinfiammatori. Inoltre, poiché la sclerite può insorgere a seguito di un’infezione o di un’allergia infettiva, nel trattamento iniziale si associano anche colliri antibiotici e antibiotici per via orale.
QCome si cura la sclerite infettiva?
A
La sclerite infettiva ha una prognosi infausta; l’identificazione dell’agente patogeno e la somministrazione precoce di antibiotici in base alla sensibilità sono essenziali. Nell’infezione da Pseudomonas aeruginosa, che progredisce rapidamente, sono necessari una potente terapia antibiotica e un rapido sbrigliamento chirurgico 7). Se la causa sono fili di sutura esposti o materiale di cerchiaggio, devono essere rimossi tempestivamente. Gli steroidi possono aggravare l’infezione, pertanto devono essere usati con cautela in caso di sospetta infezione.
La sclera è un tessuto fibroso scarsamente vascolarizzato e la sclerite profonda è rara. Tuttavia, poiché la sclera è innervata, una volta che si instaura l’infiammazione, provoca un forte dolore oculare. Lo spessore della sclera a livello dell’inserzione dei muscoli retti è di circa 0,3 mm, rendendola un sito frequente di necrosi e perforazione. La sclera non ha strutture di barriera, quindi i farmaci iniettati sotto la congiuntiva o la capsula di Tenone possono raggiungere l’interno dell’occhio per diffusione, motivo per cui le iniezioni sottocongiuntivali di steroidi sono un’opzione terapeutica.
La patologia della sclerite associata a malattie autoimmuni è caratterizzata da necrosi granulomatosa zonale. Al centro del granuloma si trova sostanza fibrinoide, circondata da cellule epitelioidi e cellule giganti multinucleate.
Nella sclerite si osserva un aumento dell’infiltrazione di cellule infiammatorie, inclusi linfociti T e macrofagi. I linfociti T e i macrofagi infiltrano il tessuto episclerale profondo e attorno ai vasi sanguigni si formano aggregati di linfociti B. L’aumentata espressione di HLA-DR e del recettore dell’IL-2 sui linfociti T suggerisce il coinvolgimento di una risposta immunitaria cellulo-mediata.
I plasmacellule sono coinvolte nella produzione di metalloproteinasi della matrice (MMP) e TNF-α. Nella sclerite necrotizzante si osserva una vasculite con necrosi fibrinoide e infiltrazione di neutrofili nella parete vascolare. Il meccanismo della sclerite endogena probabilmente coinvolge una risposta immunitaria cellulo-mediata.
Sclerite non necrotizzante (diffusa o nodulare): la vasculite non è marcata, l’infiammazione è prevalentemente non granulomatosa. Nella forma nodulare si osserva necrosi fibrinoide centrale circondata da cellule epitelioidi.
Sclerite necrotizzante: si osservano piccoli focolai di necrosi e un’infiammazione non granulomatosa composta principalmente da linfociti, plasmacellule e macrofagi. Sono caratteristiche la vasculite con necrosi fibrinoide e l’infiltrazione di neutrofili.
Sclerite infettiva: oltre all’infiammazione necrotica si formano microascessi. Nell’infezione da Nocardia, anche dopo la scomparsa dei noduli, i batteri rimangono in profondità e si verificano frequenti recidive 2).
Scleromalacia perforante: si verifica in pazienti con artrite reumatoide di lunga durata o malattie correlate. Si presenta con placche sclerali necrotiche senza congestione vicino al limbo, con un progressivo assottigliamento sclerale che porta all’esposizione dell’uvea.
È stata riportata una serie di 8 casi di sclerite posteriore insorta dopo vaccinazione o infezione da COVID-19, inizialmente scambiata per melanoma coroidale5). L’intervallo medio tra l’ultima dose di vaccino e l’insorgenza è stato di 132 giorni, e tra l’infezione da COVID-19 e l’insorgenza di 14 giorni 5). La maggior parte dei casi si è risolta spontaneamente entro 2 mesi con un impatto visivo minimo 5).
Rituximab per la sclerite refrattaria associata ad ANCA
Casi di sclerite necrotizzante associata ad ANCA resistente alla terapia immunosoppressiva convenzionale (steroidi + ciclofosfamide) hanno mostrato efficacia del rituximab (anticorpo anti-CD20) sia per l’induzione che per il mantenimento della remissione 3). Si stanno accumulando anche studi di follow-up a lungo termine sull’efficacia del rituximab nelle lesioni oculari della granulomatosi con poliangioite associata ad ANCA 3).
Sono stati riportati casi di sclerite come manifestazione oculare dell’arterite di Takayasu, che richiedono attenzione come indizio diagnostico della sindrome da aortite sistemica 4). Nelle giovani donne con sclerite necrotizzante è importante escludere l’arterite di Takayasu.
QC'è un legame con il COVID-19?
A
Sono state riportate serie di casi di sclerite posteriore dopo vaccinazione o infezione da COVID-19 5). Tuttavia, la relazione causale non è provata e la maggior parte dei casi si risolve spontaneamente. La sclerite posteriore associata a COVID-19 può essere scambiata per un tumore coroidale, quindi è importante riconoscerla come diagnosi differenziale 5).
Babu N, Kumar K, Upadhayay A, Kohli P. Nodular posterior scleritis - The great masquerader. Taiwan journal of ophthalmology. 2021;11(4):408-412. doi:10.4103/tjo.tjo_20_21. PMID:35070674; PMCID:PMC8757530.
Chauhan K, Murthy SI, Mitra S. Demystifying nocardial scleritis. BMJ case reports. 2023;16(11). doi:10.1136/bcr-2023-255730. PMID:38011958; PMCID:PMC10685915.
Tahavvori M, Fekri S, Hassanpour K, Sadoughi MM, Javadi M. Isolated ANCA-associated scleritis successfully treated with systemic rituximab; a case report and review of literature. BMC ophthalmology. 2025;25(1):176. doi:10.1186/s12886-025-04027-6. PMID:40197146; PMCID:PMC11974155.
Chittipolu S, Kennard JL, Tumma RS, Doyle AR. Scleritis in Takayasu Arteritis. Cureus. 2023;15(4):e37724. doi:10.7759/cureus.37724. PMID:37206528; PMCID:PMC10191460.
Negretti GS, Zeiger JS, Cherkas E, Shields CL. Posterior scleritis following COVID-19 vaccination or infection simulating uveal melanoma in 8 consecutive patients. Eye (London, England). 2024;38(1):185-191. doi:10.1038/s41433-023-02656-z. PMID:37422535; PMCID:PMC10764359.
Akada M, Muraoka Y, Morooka S, Ishihara K, Hata M, Tsujikawa A. SEVERE CIRCULATORY DISTURBANCE IN OPTIC DISK, RETINA, AND CHOROID AFTER SUB-TENON TRIAMCINOLONE ACETONIDE INJECTION FOR POSTERIOR SCLERITIS. Retinal cases & brief reports. 2025;19(6):789-792. doi:10.1097/ICB.0000000000001642. PMID:39058980; PMCID:PMC12570599.
Dallinga M, Murtagh P, Powell S, Murphy CC. Moraxella nonliquefaciens-associated infectious scleritis. BMJ case reports. 2023;16(5). doi:10.1136/bcr-2022-254113. PMID:37221000; PMCID:PMC10230883.