Episclerite semplice
Frequenza: più comune
Insorgenza: improvvisa
Decorso: raggiunge il picco in circa 12 ore e si risolve in 2-3 giorni
Reperti: iperemia a ventaglio (circa 67%) o diffusa (circa 33%)

L’episclerite è una malattia congestizia benigna e autolimitante del tessuto episclerale. È un’infiammazione dei plessi vascolari superficiali, come il plesso della capsula di Tenone, e rispetto alla sclerite, che colpisce vasi più profondi, il dolore è lieve e l’impatto sulla vista è minimo. Spesso è idiopatica e ricorrente, con tendenza a manifestarsi bilateralmente. L’incidenza è di 41,0 casi per 100.000 persone all’anno, con una prevalenza di 52,6.
Sebbene sia una causa relativamente frequente di arrossamento oculare, può essere confusa con congiuntivite o sclerite, portando a diagnosi errate alla prima visita. In questa malattia, lo stroma sclerale non è coinvolto e non si osserva progressione verso complicanze strutturali gravi come la perforazione oculare. Tuttavia, nei casi con decorso ricorrente o in quelli associati a malattie autoimmuni sistemiche come l’artrite reumatoide o la granulomatosi con poliangioite, è necessario trattare la malattia di base e seguire il paziente a lungo termine. Comprendere l’episclerite non solo come malattia oculare isolata, ma come ‘fenotipo oculare’ di una malattia sistemica, è fondamentale per la gestione delle recidive e il miglioramento della prognosi.
La classificazione di Watson è ampiamente utilizzata per la classificazione clinica delle malattie infiammatorie della sclera e dell’episclera. In base alla sede, si dividono in tre gruppi principali: episclerite, sclerite anteriore e sclerite posteriore. La sclerite anteriore è ulteriormente suddivisa in base alla morfologia in diffusa, nodulare e necrotizzante (infiammatoria/non infiammatoria). Una differenza importante rispetto alla sclerite anteriore è che nell’episclerite non esiste un tipo necrotizzante e morfologicamente si classifica in due tipi: semplice (tipo diffuso) e nodulare. Poiché questa classificazione riflette la profondità dell’infiammazione (superficiale o profonda) e la gravità della progressione e della prognosi, la determinazione del tipo di malattia al momento della diagnosi costituisce la base per la strategia terapeutica e la spiegazione della prognosi. L’episclerite è classificata in questo sistema come il gruppo più lieve e con la prognosi migliore.
Episclerite semplice
Frequenza: più comune
Insorgenza: improvvisa
Decorso: raggiunge il picco in circa 12 ore e si risolve in 2-3 giorni
Reperti: iperemia a ventaglio (circa 67%) o diffusa (circa 33%)
Episclerite nodulare
Frequenza: leggermente meno comune
Insorgenza: graduale
Decorso: tende a durare più a lungo rispetto alla forma semplice
Reperti: nodulo episclerale localizzato vicino al limbo corneale (mobile)
La sclera è composta da tre strati: episclera, stroma sclerale e lamina fusca. L’episclera è un tessuto connettivo vascolarizzato sopra lo stroma sclerale, compreso come una struttura fibroelastica situata tra lo stroma sclerale e la capsula di Tenone. È costituita da due strati: lo strato parietale esterno (rete capillare episclerale superficiale) e lo strato viscerale profondo (rete vascolare altamente anastomotica), entrambi originati dalle arterie ciliari anteriori. La maggior parte delle fibre nervose sono rami del nervo trigemino. L’episclera forma un plesso vascolare episclerale tra l’inserzione dei muscoli retti e il limbo, normalmente nascosto dalla congiuntiva, ma quando si infiamma si dilata causando una vivace iperemia. L’episclera si assottiglia gradualmente verso la parte posteriore dell’occhio, dove la capsula di Tenone diventa predominante.
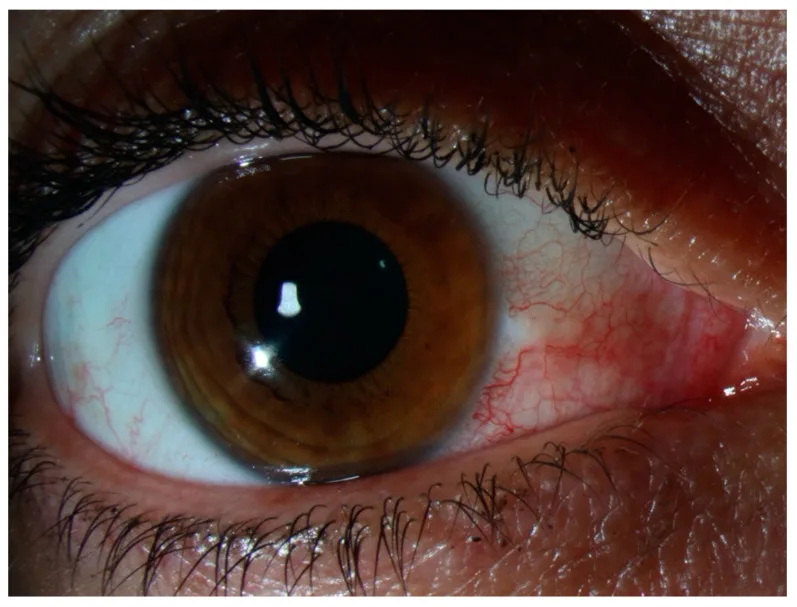
Non c’è dolorabilità alla pressione né secrezione oculare. In presenza di dolore intenso o secrezione evidente, riconsiderare sclerite, congiuntivite infettiva o uveite anteriore. I sintomi di solito si attenuano o scompaiono completamente entro pochi giorni, senza compromettere la funzione visiva. In caso di recidiva, spesso si manifesta nella stessa sede o nell’occhio controlaterale, e il paziente spesso lo riconosce come il “solito occhio rosso”. Il dolore intenso che disturba il sonno notturno, come nella sclerite, o la forte dolorabilità alla palpazione della palpebra superiore di solito non sono presenti nell’episclerite.
L’osservazione della sede e del colore dell’arrossamento è centrale per la diagnosi differenziale. L’arrossamento dell’episclerite è di colore rosso vivo o rosa, in contrasto con l’arrossamento profondo di colore rosso scuro (violaceo) della sclerite.
| Reperto | Episclerite | Sclerite |
|---|---|---|
| Colore dell’arrossamento | Rosso vivo o rosa | Rosso scuro (violaceo) |
| Dolore | Lieve o assente | Forte e irradiato |
| Mobilità del nodulo | Presente | Assente |
L’acuità visiva è generalmente normale. Chemosi, ipertensione oculare, uveite anteriore e cheratite sono rare; se presenti, considerare sclerite o altre patologie. L’assenza di infiammazione della congiuntiva tarsale aiuta a distinguerla dalla congiuntivite. Nella sclerite, l’infiammazione può estendersi ai tessuti circostanti causando infiltrati corneali periferici, ulcere o uveite anteriore, mentre l’episclerite è autolimitante e raramente coinvolge i tessuti adiacenti. Alla lampada a fessura, identificare il livello del plesso vascolare sclerale; se una lesione rialzata rossa non permette di visualizzare i vasi sclerali, considerare una lesione neoplastica.
L’episclerite è caratterizzata da assenza di secrezione e iperemia localizzata vicino al limbo corneale. La congiuntivite di solito è indolore con secrezione, e l’iperemia è più marcata nel fornice, diminuendo verso il limbo. Alla lampada a fessura, i vasi episclerali non sono mobili, mentre quelli congiuntivali lo sono. Per dettagli, vedere la sezione Diagnosi e metodi di esame.
La maggior parte dei casi è idiopatica (causa sconosciuta); circa il 26-36% dei casi è associato a malattie sistemiche. Anche nei casi idiopatici, si sospetta un coinvolgimento immunologico, con una reazione infiammatoria aspecifica a predominanza linfocitaria nel plesso vascolare episclerale superficiale. Il decorso ricorrente e la tendenza alla bilateralità suggeriscono una disregolazione immunitaria sistemica di base.
Malattie del collageno e autoimmuni (più comune: artrite reumatoide)1):
Vasculite:
Infezioni: batteri, micobatteri, sifilide, malattia di Lyme, virus herpes, herpes zoster possono esserne causa. L’episclerite associata a herpes zoster oftalmico è considerata una risposta immunitaria all’agente patogeno piuttosto che un’infezione diretta. È stato riportato anche un caso di parassitosi sottocongiuntivale da Dirofilaria repens erroneamente diagnosticata come episclerite7).
Altro: gotta, atopia, corpi estranei, traumi chimici, farmaci (topiramato, pamidronato), e segnalazioni come sintomo iniziale di COVID-19.
Sì, circa il 30% dei pazienti presenta una malattia sistemica associata. La più comune è l’artrite reumatoide, ma può anche essere il sintomo iniziale di malattie come la granulomatosi con poliangioite (GPA) o la malattia di Behçet, la cui diagnosi e trattamento precoci influenzano la prognosi. In caso di recidive frequenti o sintomi sistemici, si raccomanda una valutazione sistemica con fattore reumatoide, anticorpi antinucleo, ANCA ed esame delle urine.
L’episclerite viene diagnosticata principalmente in base all’anamnesi e all’esame con lampada a fessura. È fondamentale osservare attentamente con la lampada a fessura il livello dei vasi episclerali (superficiale o profondo), il colore dell’arrossamento, la presenza di noduli e l’eventuale assottigliamento o necrosi.
La fenilefrina al 2,5% in collirio contrae i vasi congiuntivali ed è utile per distinguere la congiuntivite dall’episclerite. La fenilefrina al 10% contrae i vasi episclerali superficiali ma non quelli profondi, consentendo di differenziare l’episclerite dalla sclerite.
Il test di reazione con epinefrina in collirio alla diluizione 1:1000 è un metodo semplice per valutare il coinvolgimento dei vasi profondi. Se l’arrossamento scompare dopo l’instillazione, si tratta di episclerite; se non scompare, indica sclerite. La valutazione complessiva si basa su tre elementi: numero e mobilità dei noduli, presenza di dolore/dolorabilità e reazione all’epinefrina.
I test di reazione con epinefrina e fenilefrina sono particolarmente utili come ausilio diagnostico quando non è possibile visualizzare direttamente la stratificazione dell’arrossamento con la lampada a fessura o in caso di noduli piccoli. L’osservazione a 10-15 minuti dall’instillazione permette di valutare la contrazione dei vasi superficiali; se persiste l’arrossamento dei vasi profondi, si dà priorità al trattamento per la sclerite.
La tenonite è considerata un tipo di episclerite e la loro differenziazione clinica è difficile. La diagnosi si basa sulla combinazione di mobilità del nodulo, presenza di dolore/dolorabilità, risposta alla fenilefrina topica e colorazione con fluoresceina.
Per episclerite singola e lieve non è necessaria un’ampia indagine sistemica. In caso di recidive frequenti o sintomi sistemici associati, considerare i seguenti esami.
Nei casi in cui l’episclerite si presenta come manifestazione iniziale della granulomatosi con poliangioite, può coesistere una compromissione della funzionalità renale 3). Se sono presenti sia infiammazione oculare che anomalie della funzionalità renale, è necessario eseguire tempestivamente una ricerca di vasculite sistemica, inclusa la granulomatosi con poliangioite. Nell’episclerite refrattaria o ricorrente, è consigliabile valutare l’attività della malattia e avviare il trattamento della malattia di base in collaborazione con reumatologi e internisti.
Oltre alla valutazione con lampada a fessura, la tomografia a coerenza ottica del segmento anteriore (AS-OCT) per valutare lo spessore dello strato episclerale e il decorso vascolare, e l’ecografia (modalità B) per valutare lo spessore sclerale possono essere utilizzate come diagnostica complementare. Per escludere la sclerite necrotizzante o valutare la presenza di sclerite posteriore, l’ecografia verifica la presenza del segno T (accumulo di liquido intorno alla guaina del nervo ottico). Nell’episclerite comune, questi esami di imaging spesso non mostrano reperti specifici, e la diagnosi si basa sulla combinazione dell’esame diretto con lampada a fessura, dell’anamnesi e della valutazione sistemica.
L’episclerite spesso guarisce spontaneamente in pochi giorni o settimane senza trattamento. Spiegare al paziente la natura benigna della malattia, il decorso spontaneo e la necessità di una valutazione per malattie sistemiche, oltre a fornire rassicurazione, è il primo passo nella gestione. L’applicazione di impacchi freddi o lacrime artificiali refrigerate è efficace per ridurre sintomi soggettivi come irritazione e sensazione di calore. Nei casi lievi, si evita un intervento farmacologico aggressivo e si conferma la risoluzione spontanea con follow-up a breve termine (giorni), evitando così rebound ed effetti collaterali legati al trattamento.
I colliri steroidei a bassa concentrazione sono la prima scelta. Spesso si associa un collirio antibiotico per la diagnosi differenziale con la sclerite.
Se la risposta al trattamento con colliri è scarsa, si considera il passaggio all’esame e al trattamento per la sclerite. Sebbene i colliri steroidei sopprimano rapidamente i sintomi, l’uso prolungato o ripetuto può aumentare il rischio di recidiva e indurre arrossamento da ‘rimbalzo’.
Il trattamento prevede una riduzione graduale e la sospensione dopo la risoluzione dei sintomi, evitando una somministrazione prolungata indiscriminata. L’uso a lungo termine di colliri steroidei comporta il rischio di aumento della pressione intraoculare steroideo-dipendente e cataratta sottocapsulare posteriore; pertanto, dopo 1-2 settimane, si verifica il miglioramento e si riduce gradualmente. In caso di recidive, si valuta individualmente l’attività della malattia in ogni episodio e si dà priorità all’ottimizzazione del trattamento della malattia sistemica di base.
Nell’episclerite associata a malattie del collagene come l’artrite reumatoide, il trattamento della malattia di base è direttamente correlato alla prognosi1). Se la terapia locale è resistente, si associa prednisolone per via orale (terapia a dosi decrescenti da 20-30 mg/die). Salvo chiara evidenza di concomitante malattia infiammatoria sistemica, i casi che richiedono terapia steroidea sistemica sono molto rari.
Nell’episclerite associata a granulomatosi con poliangioite, la terapia di induzione della remissione con ciclofosfamide o rituximab è efficace3)4). Il rituximab ha mostrato un tasso di remissione a 6 mesi più elevato rispetto alla ciclofosfamide (64% vs 53%) secondo alcuni studi3).
I colliri steroidei sopprimono rapidamente i sintomi dell’episclerite, ma è stato segnalato che la loro sospensione può causare arrossamento da ‘effetto rebound’, portando a una riacutizzazione più grave. Pertanto, l’uso di steroidi è dibattuto; nei casi lievi, alcuni preferiscono l’osservazione senza trattamento o i FANS. In caso di recidive frequenti, si raccomanda l’assunzione orale di inibitori della COX2 o l’approfondimento delle malattie sistemiche.
Il meccanismo di insorgenza dell’episclerite non è ancora completamente chiarito. Nelle aree colpite si osservano vasodilatazione e iperemia del plesso vascolare episclerale superficiale, con infiltrazione di cellule infiammatorie, prevalentemente linfociti, nell’episclera e nella capsula di Tenone. La differenza sostanziale rispetto alla sclerite è che il parenchima sclerale stesso non è coinvolto. L’infiltrato infiammatorio è composto principalmente da linfociti T e da un piccolo numero di plasmacellule; non si osservano solitamente immagini di infiammazione purulenta a predominanza neutrofila né formazione di granulomi.
Istologicamente si tratta di un’infiammazione non granulomatosa, caratterizzata principalmente da vasodilatazione e infiltrazione linfocitaria. Nell’episclerite nodulare, al centro della lesione si osserva necrosi fibrinoide circondata da un disposizione di cellule epitelioidi. Questi reperti sono simili a quelli dell’infiammazione granulomatosa osservata nella sclerite, e alcuni autori considerano episclerite e sclerite come uno spettro di malattia basato sulla profondità dell’infiammazione. La necrosi fibrinoide su piccola scala osservata nell’episclerite può essere interpretata come la forma lieve dei cambiamenti necrotici più estesi della sclerite.
La progressione dell’infiammazione aumenta la produzione di specie reattive dell’ossigeno (ROS) e potenzia lo stress ossidativo2). Il contenuto totale di vitamina C nella retina umana è circa 20 volte superiore a quello plasmatico, e i tessuti oculari dipendono fortemente dal sistema antiossidante. Nell’episclerite autoimmune, si ipotizza che una ridotta funzionalità di questo sistema antiossidante possa causare infiammazione cronica e danno tissutale dell’episclera2). I ROS danneggiano l’endotelio vascolare e inducono il rilascio di citochine infiammatorie, provocando vasodilatazione persistente e aumento della permeabilità. L’esposizione cronica allo stress ossidativo della superficie oculare e dell’episclera è considerata un fattore contribuente all’episclerite ricorrente, e si sta valutando il significato terapeutico di un intervento antiossidante.
Clinicamente, l’episclerite raramente evolve direttamente in sclerite. D’altra parte, nella maggior parte dei casi di sclerite si osserva anche infiammazione dell’episclera (cambiamenti episcleritici), per cui le due condizioni sono intese più come un continuum basato sulla profondità dello strato vascolare coinvolto che come malattie completamente indipendenti. L’episclerite colpisce principalmente il plesso vascolare episclerale superficiale (strato parietale), mentre la sclerite coinvolge il plesso vascolare profondo e il parenchima sclerale.
È noto che a livello dell’inserzione dei muscoli retti lo spessore della sclera è di circa 0,3 mm, il più sottile, rendendola più vulnerabile a infiammazione e traumi. Il plesso vascolare episclerale riceve un ricco apporto sanguigno attraverso le arterie ciliari anteriori, quindi l’iperemia tende a manifestarsi rapidamente durante l’infiammazione. D’altra parte, la sclera stessa è un tessuto povero di vasi, e l’infiammazione profonda come la sclerite è rara. La caratteristica anatomica per cui i vasi derivati dalle arterie ciliari anteriori nell’episclerite sono reversibilmente congesti è la base meccanicistica della rapida risoluzione dell’iperemia con il test alla fenilefrina; l’assenza di questa reazione nella sclerite profonda costituisce un criterio fisiopatologico per la diagnosi differenziale.
Esiste un case report su un uomo di 60 anni con episclerite idiopatica recidivante, in cui l’inizio di vitamina C 500 mg/die per via orale ha portato a un’assenza di recidive per 7 mesi2). La vitamina C è un potente antiossidante e si ipotizza che possa sopprimere l’infiammazione dei tessuti oculari riducendo lo stress ossidativo. È noto che i tessuti oculari dipendono fortemente dal sistema antiossidante, con concentrazioni di vitamina C nella retina circa 20 volte superiori a quelle plasmatiche; pertanto, l’integrazione di vitamina C e altri nutrienti antiossidanti potrebbe rappresentare una strategia candidata per la prevenzione delle recidive2). Tuttavia, per stabilirne l’efficacia sono necessari studi caso-controllo e studi clinici con gruppo di controllo2). Allo stato attuale, questa opzione è considerata solo come supporto in casi di recidive con sintomi gravi o in presenza di secchezza oculare e infiammazione cronica della superficie oculare.
La granulomatosi con poliangioite (GPA) è una malattia fatale con un tasso di mortalità a 1 anno dell’80% se non trattata, ma l’introduzione della terapia immunosoppressiva può ridurre la mortalità al 10%3). Poiché l’episclerite può essere il sintomo iniziale della GPA, gli oftalmologi devono riconoscere questa associazione e, in caso di episclerite recidivante, eseguire attivamente esami sistemici3)4). In particolare, la coesistenza di infiammazione oculare e disfunzione renale è un reperto fortemente suggestivo di granulomatosi con poliangioite3).
Per l’episclerite e la sclerite associate all’artrite reumatoide, è stata riportata l’efficacia di farmaci biologici come gli inibitori del TNFα e il rituximab1). Infliximab e adalimumab hanno dimostrato efficacia nell’artrite reumatoide e nell’uveite, e vengono considerati anche per la sclerite e l’episclerite refrattarie. D’altra parte, l’etanercept è noto per una reazione paradossa che induce o aggrava l’infiammazione oculare, richiedendo cautela nella scelta del farmaco1). Il rituximab è un anticorpo monoclonale diretto contro le cellule B e ha mostrato efficacia nell’infiammazione oculare associata a vasculite. L’uso di questi farmaci biologici viene deciso in stretta collaborazione con i reumatologi e gli internisti specializzati in malattie del collagene.
Sono stati riportati casi in cui pazienti diagnosticati con episclerite presentavano in realtà un tumore metastatico intraoculare 6) o una parassitosi sottocongiuntivale 7); pertanto, nell’episclerite refrattaria e ricorrente è importante escludere malattie maligne o infezioni. Gli esami di imaging e la valutazione dettagliata dei reperti biomicroscopici di masse contenenti vasi forniscono indizi diagnostici. È necessario valutare complessivamente la mobilità della massa, la trasparenza dei vasi sclerali, la presenza di aderenze ai tessuti circostanti e la risposta al trattamento; lesioni rilevate persistenti che non rispondono ai normali colliri steroidei costituiscono un motivo per considerare attivamente una biopsia o un approfondimento con imaging.
Gli studi osservazionali a lungo termine sul decorso naturale dell’episclerite e sul tempo di manifestazione delle malattie sistemiche sono limitati, e in particolare i dati sull’incidenza e sul profilo delle malattie associate nella popolazione giapponese non sono sufficienti. I dati aggregati precedentemente riportati dall’Europa e dagli Stati Uniti mostrano un’incidenza annuale di circa 40-60 casi per 100.000 persone, ma i valori variano a causa di differenze etniche, ambientali e nella gestione dei registri delle uveiti. La futura creazione di registri clinici e studi multicentrici potrebbe consentire l’identificazione dei fattori di rischio di recidiva e della tempistica di manifestazione delle malattie sistemiche.
- Promelle V, Goeb V, Gueudry J. Rheumatoid Arthritis Associated Episcleritis and Scleritis: An Update on Treatment Perspectives. Journal of clinical medicine. 2021;10(10). doi:10.3390/jcm10102118. PMID:34068884; PMCID:PMC8156434.
- Goyal L, Ajmera K, Pandit R. Reoccurring Episcleritis and the Role of Antioxidants. Cureus. 2022;14(4):e24111. doi:10.7759/cureus.24111. PMID:35530867; PMCID:PMC9073074.
- Foster LD, Nyugen M, Margolin E. Conjunctivitis, episcleritis and anterior uveitis as the first presenting features of granulomatosis with polyangiitis. BMJ Case Rep. 2021;14:e243558. doi:10.1136/bcr-2021-243558.
- Ciotoracu AC, Dimăncescu MG, Mitulescu TC, et al. A clinical case of recurrent episcleritis as the initial manifestation of granulomatosis with polyangiitis. Rom J Ophthalmol. 2021;65(4):386-390. doi:10.22336/rjo.2021.76.
- Jari M, Nasiri S, Ghandehari M. Episcleritis and posterior uveitis misdiagnosed as orbital cellulitis in a child patient with Behçet’s disease. SAGE Open Med Case Rep. 2023;11:1-4. doi:10.1177/2050313x231182237.
- Chong YJ, Azzopardi M, Ng B, Salvi SM, Sreekantam S. Ocular Metastasis as First Presentation of Large-Cell Neuroendocrine Carcinoma. Case reports in ophthalmology. 2023;14(1):684-691. doi:10.1159/000535233. PMID:38090108; PMCID:PMC10715755.
- Redón-Soriano M, Blasco A, Gomila B, González-Sánchez M, Simón F, Esteban JG. Subconjunctival human dirofilariasis by Dirofilaria repens in the Mediterranean Basin. American journal of ophthalmology case reports. 2022;26:101570. doi:10.1016/j.ajoc.2022.101570. PMID:35586152; PMCID:PMC9108447.