Il linfoma maligno congiuntivale è un linfoma maligno primitivo della congiuntiva dovuto alla proliferazione monoclonale di cellule B. Origina dal tessuto linfoide associato alla congiuntiva (CALT) e rappresenta il 25-30% dei linfomi degli annessi oculari. 1)
La maggior parte di essi corrisponde al linfoma MALT (linfoma del tessuto linfoide associato alle mucose). Il linfoma MALT primitivo della congiuntiva è anche chiamato linfoma CALT, con tendenza a bassa malignità, progressione lenta e localizzazione mucosa, distinguendosi nettamente per natura e prognosi dagli altri linfomi ad alta malignità. D’altra parte, i linfomi diffusi a cellule medie e grandi presentano numerose figure mitotiche e sono più probabilmente metastasi di linfomi di altri organi.
L’incidenza è rara, con 0,2 casi per 100.000 persone, corrispondente all’1-2% di tutti i linfomi non Hodgkin. 1) Si verifica principalmente dopo i 60 anni e l’incidenza del linfoma della zona marginale extranodale e del linfoma follicolare ha mostrato un trend in aumento tra il 1980 e il 2005. 1) Sebbene più comune negli anziani, sono stati riportati casi a 33 mesi di età. L’incidenza del linfoma degli annessi oculari nei bambini è estremamente rara, con 0,12 casi per milione di persone. 1)
I sottotipi istologici, secondo una raccolta di 1014 casi, sono: linfoma della zona marginale extranodale 81%, linfoma follicolare (FL) 8%, linfoma a cellule del mantello (MCL) 3%, linfoma diffuso a grandi cellule B (DLBCL) 3%, e il 98% è di tipo B. 1) La maggior parte sono linfomi B non Hodgkin, in linea con la tendenza generale dei linfomi in ambito oftalmologico.
Storicamente, Arnold e Becker riportarono il primo caso nel 1872, e Isaacson stabilì il concetto di tessuto linfoide associato alle mucose (MALT) nel 1984. 1) La classificazione OMS 2017 definisce oltre 80 sottotipi. 1)
QQual è la frequenza del linfoma maligno congiuntivale?
A
L’incidenza è di 0,2 casi per 100.000 persone, rappresentando il 25-30% di tutti i linfomi degli annessi oculari. 1) È una malattia rara, corrispondente all’1-2% di tutti i linfomi non Hodgkin. Si verifica principalmente dopo i 60 anni, ed è estremamente rara nei bambini, con 0,12 casi per milione di bambini. 1)
QCos'è il linfoma MALT?
A
Il linfoma MALT (linfoma del tessuto linfoide associato alle mucose) è un termine generico per i linfomi B di basso grado che originano da tessuti mucosi come tratto gastrointestinale, ghiandole salivari, polmoni, orbita e congiuntiva. Origina dal tessuto linfoide acquisito localmente a causa di infiammazione cronica o infezione, e si caratterizza per progressione lenta e limitazione locale. Quelli originati dalla congiuntiva sono chiamati linfomi CALT e hanno una prognosi relativamente favorevole.
McGrath LA, et al. Conjunctival Lymphoma. Eye (Lond). 2023. Figure 5. PMCID: PMC10049989. License: CC BY.
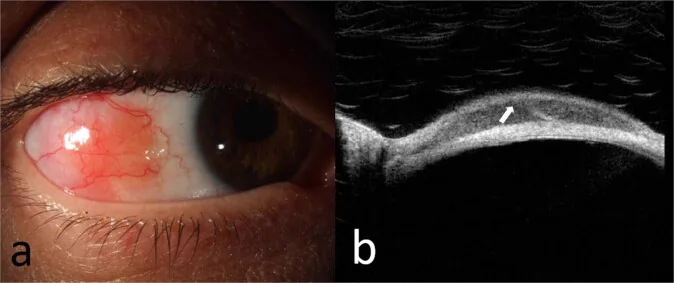
Fotografia con lampada a fessura (a) di un linfoma della zona marginale extranodale della congiuntiva bulbare e OCT (b) della lesione che mostra un infiltrato subepiteliale omogeneo iporiflettente e una banda subepiteliale iperriflettente. Ciò corrisponde alle lesioni congiuntivali trattate nella sezione «2. Principali sintomi e segni clinici».
Il sintomo principale è una massa congiuntivale, spesso con scarsa sensazione di corpo estraneo. In alcuni casi la diagnosi viene posta dopo un lungo trattamento come congiuntivite cronica.
Massa/rilievo: percepita come una massa congiuntivale che aumenta lentamente.
Sensazione di corpo estraneo/lieve disagio: l’85% presenta qualche sintomo, di cui il 67% lieve. 1)
Ptosi: può verificarsi a causa dell’aumento della massa.
Intervallo tra comparsa dei sintomi e consultazione: nei linfomi di basso grado, i sintomi sono spesso presenti da 4-6 mesi.
Reperti clinici (segni riscontrati dal medico durante la visita)
Lesioni rilevate, traslucide, di colore rosa salmone compaiono sulla congiuntiva del fornice e bulbare. È difficile distinguerle visivamente dall’iperplasia linfoide reattiva (RLH).
La sede preferita è la congiuntiva del fornice e, progredendo, può estendersi a tutta la congiuntiva. È importante anche esaminare la congiuntiva della palpebra superiore rovesciandola. Oltre il 90% dei casi si presenta di colore rosa, con diametro di base tipico di 15 mm, spessore di 3 mm e 2-3 lesioni per occhio. 1) I due terzi dei casi sono unilaterali (linfoma della zona marginale extranodale nell’82%), il 62% si verifica nella congiuntiva superiore o inferiore. Solo il 7% raggiunge il limbo. 1) Può essere bilaterale, manifestandosi contemporaneamente o dopo anni nell’occhio controlaterale.
Le caratteristiche cliniche per sottotipo sono le seguenti:
Linfoma della zona marginale extranodale
Aspetto : lesione maculare piatta, mobile, di colore rosa salmone.
Distribuzione : prevalentemente a fornice e congiuntiva bulbare. Spesso unilaterale (82%). 1)
Caratteristiche : di basso grado, progressione lenta. Sottotipo più comune (81%). Prognosi favorevole.
Linfoma follicolare / a cellule del mantello
Linfoma follicolare : si presenta spesso come lesioni multinodulari. 1)
Linfoma a cellule del mantello : massa voluminosa, rosso scuro. 1)
Caratteristiche : il linfoma follicolare è di basso grado. Il linfoma a cellule del mantello è spesso associato a malattia sistemica.
Linfoma diffuso a grandi cellule B / a cellule T
Linfoma diffuso a grandi cellule B : massa grigiastra a rapida crescita. 1)
Linfoma a cellule T : il 30% si verifica al limbo. Alcuni casi si accompagnano a sclerite. 1)
Caratteristiche: Alto grado. È necessario un intervento urgente.
QMassa color salmone = linfoma?
A
Una massa congiuntivale color salmone può presentarsi anche nell’iperplasia linfoide reattiva. La differenziazione basata solo sull’aspetto è difficile; per la diagnosi definitiva è necessaria una biopsia. Per i dettagli, vedere la sezione «Diagnosi e metodi di esame».
Il meccanismo di sviluppo del linfoma maligno congiuntivale è considerato una disregolazione delle cellule B dovuta a stimolazione antigenica cronica. Normalmente non c’è tessuto linfoide nella congiuntiva, ma un’infiammazione cronica persistente può portare all’acquisizione di tessuto linfoide associato alle mucose, da cui si sviluppa un linfoma MALT.
Agenti infettivi: Sono state riportate associazioni con Helicobacter pylori, Chlamydophila psittaci, virus dell’epatite C e HIV. 1) L’associazione con C. psittaci è più frequentemente riportata in Europa e il tasso di positività varia a seconda della regione.
Malattie autoimmuni: Associazione con sindrome di Sjögren, tiroidite di Hashimoto e malattie correlate a IgG4. 1)
Microbiota congiuntivale: Delftia sp. (bacillo Gram-negativo) potrebbe essere coinvolto. 1)
Relazione con l’iperplasia linfoide reattiva: L’iperplasia linfoide reattiva è considerata una condizione precursore. 1)
Per la diagnosi definitiva è indispensabile l’esame istopatologico tramite biopsia. La diagnosi non può essere posta solo sulla base dei reperti clinici e di imaging. Il linfoma maligno è monoclonale, mentre l’iperplasia linfoide reattiva è policlonale; questo è il punto centrale della differenziazione.
La biopsia consiste nell’asportazione di una parte o di tutta la neoplasia per l’esame patologico. Si raccomanda di ottenere un campione bioptico di almeno 250 mg (necessario per i test di riarrangiamento genico). Poiché il linfoma congiuntivale cresce in modo sottile e diffuso, può essere necessario dividere l’area di resezione in due siti per evitare la simblefaron.
Colorazione HE + immunoistochimica : confermare la linea di appartenenza linfocitaria e la clonalità. Utilizzare CD10, CD20, CD79a, CD5, ecc.
Citometria a flusso : eseguibile se la quantità di campione è sufficiente. Consente di determinare rapidamente la linea e il grado di differenziazione delle cellule tumorali.
Analisi del riarrangiamento genico : mediante Southern blot o PCR per verificare la presenza di riarrangiamenti dei geni delle immunoglobuline/recettori delle cellule T. Nel linfoma maligno, un pattern di riarrangiamento monoclonale è identico in tutte le cellule tumorali, distinguendolo dall’iperplasia linfoide reattiva policlonale.
I profili immunologici per sottotipo sono i seguenti:
Sottotipo
Marcatori caratteristici
Linfoma della zona marginale extranodale (EMZL / MALT)
Una volta diagnosticato un linfoma maligno, si effettua una ricerca sistemica per individuare eventuali lesioni primitive. È importante eseguire la stadiazione in collaborazione con un ematologo.
La ricerca sistemica comprende quanto segue:
FDG-PET/TC : utilizzata per la stadiazione sistemica. 1)
Biopsia del midollo osseo : valuta la presenza di disseminazione sistemica.
LDH sierico e β2-microglobulina : utilizzati come marcatori tumorali di riferimento.
Lo stadio viene valutato secondo la classificazione di Ann Arbor (per il linfoma delle appendici oculari può essere applicata anche la classificazione di Lugano) e determina direttamente la strategia terapeutica.
Stadio
Definizione
Indicazione terapeutica
Stadio IE (localizzato)
Limitato alla congiuntiva o alle appendici oculari
Radioterapia come prima scelta
Stadio IIE o superiore
Coinvolgimento linfonodale o di altri organi
Considerare chemioterapia sistemica
Esami di imaging ausiliari e diagnosi differenziale
Tomografia a coerenza ottica ad alta risoluzione (HR-OCT): Può essere utilizzata per una valutazione ausiliaria, ma la diagnosi definitiva è solo patologica. 1)
Diagnosi differenziali: È necessario differenziare da iperplasia linfoide reattiva, malattia oculare associata a IgG4, congiuntivite cronica, pterigio, granuloma piogenico, ecc. 1)
QÈ possibile diagnosticare un linfoma congiuntivale senza biopsia?
A
La tomografia a coerenza ottica ad alta risoluzione fornisce informazioni ausiliarie, ma la diagnosi definitiva è possibile solo tramite esame istopatologico di una biopsia. 1) La PET/TC è utile per la stadiazione sistemica, ma non viene utilizzata per la diagnosi locale. La conferma della monoclonalità tramite analisi del riarrangiamento genico (PCR o Southern blot) è la base per la diagnosi definitiva.
Il trattamento viene effettuato in collaborazione tra ematologia e oftalmologia. La resezione chirurgica completa è difficile; il tumore è radiosensibile e risponde bene alla chemioterapia. La strategia terapeutica è determinata in base allo stadio (classificazione di Ann Arbor) e al sottotipo istologico.
In caso di sospetta iperplasia linfoide reattiva, si può prima osservare l’evoluzione con collirio di ciclosporina A allo 0,05% (2-4 volte al giorno) o con collirio steroideo a bassa concentrazione. A volte si ottiene una regressione, ma se viene diagnosticata una malignità, si passa rapidamente al trattamento successivo.
Trattamento dei casi localizzati (stadio IE di Ann Arbor)
La radioterapia è la prima scelta. È particolarmente efficace nel linfoma MALT. Durante l’irradiazione, l’occhio viene protetto con una lente a contatto in piombo. Può essere utilizzata anche l’irradiazione con fasci di elettroni.
La radioterapia esterna con 24 Gy in 12 frazioni ha mostrato un tasso di controllo locale a 5 anni dell’89-100%. 1) L’uso di irradiazione a dose ultrabassa (4 Gy in 2 frazioni) per ridurre gli effetti collaterali ha ottenuto un tasso di risposta completa dell’85% e un tasso di controllo a 2 anni del 75%. 1)
La monoterapia con rituximab (375 mg/m² × 4 cicli) è un’opzione. 1) L’iniezione locale intralesionale di rituximab (50 mg) viene utilizzata nei casi di recidiva o in pazienti che desiderano un trattamento locale, con un tasso di risposta completa riportato del 73%. 1)
Per i linfomi CD20-positivi si sceglie la terapia R-CHOP (rituximab 375 mg/m² + CHOP). L’interferone alfa-2b (1-1,5 MUI per via sottocutanea 3 volte a settimana) ha mostrato un tasso di sopravvivenza libera da progressione a 5 anni dell’85% nei casi a basso grado e localizzati. 1)
Nei casi positivi alla PCR per Chlamydophila psittaci, è stato tentato un trattamento antibiotico con doxiciclina 100 mg (2 volte al giorno), con un tasso di sopravvivenza libera da progressione a 5 anni riportato del 55%. 1)
I dati di efficacia di ciascun trattamento sono mostrati di seguito.
Trattamento
Indicazioni principali
Tasso di risposta principale
Radioterapia esterna 24 Gy/12 frazioni
Prima scelta per casi localizzati
Controllo a 5 anni 89–100%1)
Radioterapia esterna a dose ultra-bassa 4 Gy/2 frazioni
Casi che desiderano ridurre gli effetti collaterali
Sopravvivenza libera da progressione a 5 anni 55%1)
Lo studio IELSG-19 ha mostrato una sopravvivenza libera da progressione a 5 anni del 68% vs 51% e un tasso di risposta del 95% vs 86% per il gruppo rituximab + clorambucile rispetto al gruppo clorambucile da solo.1) La crioterapia ha riportato una regressione delle lesioni nel 98% di 42 casi.1)
Per il linfoma linfoblastico a cellule T, con la terapia hyper-CVAD + HD-MA è stata riportata una risposta completa del 91%, una sopravvivenza libera da progressione a 3 anni del 66% e una sopravvivenza globale a 3 anni del 70%.2)
QPer quanto tempo è necessario il follow-up dopo il trattamento?
A
Il linfoma della zona marginale extranodale dopo il trattamento progredisce verso una malattia sistemica in circa il 20% dei casi e talvolta si osservano casi di disseminazione dopo oltre 10 anni.1) Per la diagnosi precoce di recidiva o progressione sistemica è necessario un follow-up regolare a lungo termine. La frequenza e la durata del follow-up devono essere stabilite con il medico curante.
6. Fisiopatologia e meccanismi dettagliati della malattia
Il linfoma maligno congiuntivale origina dal tessuto linfoide associato alla congiuntiva (situato al limbo e al fornice). Normalmente la congiuntiva non contiene tessuto linfoide, ma l’infiammazione cronica, l’infezione o la stimolazione autoimmune possono portare all’acquisizione di tessuto linfoide associato alle mucose, in cui si verifica la trasformazione linfomatosa.
Meccanismi molecolari del linfoma della zona marginale extranodale (linfoma MALT)
Il linfoma MALT deriva dalla proliferazione tumorale delle cellule della zona marginale (cellule B) attraverso l’attivazione cronica della via NF-κB.1)
Inattivazione del gene A20: osservata in circa il 30% dei casi, deregola la via NF-κB ed è un fattore prognostico sfavorevole. 1)
Mutazione MYD88: osservata in circa il 7% dei casi, associata a prognosi sfavorevole. 1)
Traslocazioni cromosomiche: t(14;18) 0–38%, t(11;18) 0–15%, t(3;14) 0–14% sono state riportate. 1)
Anomalie del numero di copie: si osservano trisomie 3, 12 e 18; l’anomalia del cromosoma 6 è considerata specifica degli annessi oculari. 1)
Per determinare se una proliferazione è neoplastica (maligna) o reattiva (benigna), è utile l’analisi del riarrangiamento dei geni delle immunoglobuline/recettori delle cellule T. Se viene confermata una proliferazione monoclonale, viene considerata maligna; se policlonale, viene trattata come reattiva. Anche se i reperti clinici sono simili, questo test di biologia molecolare è essenziale per la diagnosi definitiva.
Sugawara et al. (2022) hanno riportato un caso estremamente raro di linfoma linfoblastico a cellule T precursore della congiuntiva. Un uomo di 61 anni presentava una massa congiuntivale color salmone, con immunofenotipo CD7+, CD10+, TdT+, CD20−. La sopravvivenza globale a 5 anni negli adulti con linfoma linfoblastico a cellule T, che rappresenta circa il 2% di tutti i linfomi, è del 26%, risultando sfavorevole. 2)
L’analisi completa delle mutazioni genetiche mediante sequenziamento di nuova generazione sta portando all’identificazione di marcatori prognostici. 1) L’espressione di BCL-6, MUM1/IRF4 e Ki-67, dimensioni tumorali >30 mm e Ki-67 >10% sono considerati fattori prognostici sfavorevoli. 1)
Monitoraggio terapeutico mediante tomografia a coerenza ottica ad alta risoluzione
È in fase di studio l’utilità del monitoraggio non invasivo dell’efficacia terapeutica mediante tomografia a coerenza ottica ad alta risoluzione. 1) Ciò potrebbe consentire di seguire i cambiamenti tumorali senza ripetute biopsie.
Microbiota congiuntivale e meccanismo patogenetico
È stata suggerita un’associazione tra il microbiota congiuntivale, incluso Delftia sp. (bacillo Gram-negativo), e lo sviluppo del linfoma congiuntivale, e la ricerca è in corso su questa nuova via di stimolazione antigenica cronica. 1)
È in fase di studio l’applicazione di farmaci a bersaglio molecolare, inclusi gli inibitori di BTK, al linfoma congiuntivale. 1) Sono considerati una nuova opzione terapeutica per i casi recidivanti o refrattari.
McGrath LA, Ryan DA, Warrier SK, Coupland SE, Glasson WJ. Conjunctival Lymphoma. Eye. 2023;37:837-848.
Sugawara R, Usui Y, Takahashi R, Nagao T, Goto H.. A case of conjunctival precursor T cell lymphoblastic lymphoma presenting with salmon colored conjunctival mass. Am J Ophthalmol Case Rep. 2022;25:101382. doi:10.1016/j.ajoc.2022.101382. PMID:35243143; PMCID:PMC8859792.
Baltă AC, Mihai MA, Ionescu AM, Radu M, Chițac I, Murgoi G, et al. Conjunctival lymphoma: case report. Rom J Ophthalmol. 2025;69(3):440-449. PMID: 41189780.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.