El linfoma maligno conjuntival es un linfoma maligno primario de la conjuntiva debido a la proliferación monoclonal de células B. Se origina en el tejido linfoide asociado a la conjuntiva (CALT) y representa el 25–30% de los linfomas del anexo ocular. 1)
La mayoría de estos casos corresponden al linfoma MALT (linfoma del tejido linfoide asociado a mucosas). El linfoma MALT conjuntival primario también se denomina linfoma CALT y tiende a ser de bajo grado, de progresión lenta y localizado en la mucosa, lo que difiere mucho en naturaleza y pronóstico de otros linfomas de alto grado. Por el contrario, los linfomas difusos de células medianas y grandes tienen muchas figuras mitóticas y es probable que sean metástasis de linfomas de otros órganos.
La incidencia es rara, con 0,2 casos por cada 100.000 personas, lo que representa el 1–2% de todos los linfomas no Hodgkin. 1) Es más común después de los 60 años, y la incidencia del linfoma de la zona marginal extranodal y el linfoma folicular mostró una tendencia creciente entre 1980 y 2005. 1) Aunque es más común en ancianos, se han reportado casos a los 33 meses de edad. La incidencia del linfoma del anexo ocular en niños es extremadamente rara, con 0,12 casos por millón. 1)
Los subtipos histológicos en una serie de 1014 casos son: linfoma de la zona marginal extranodal 81%, linfoma folicular (FL) 8%, linfoma de células del manto (MCL) 3%, linfoma difuso de células B grandes (DLBCL) 3%, con un 98% de estirpe de células B. 1) La mayoría son linfomas no Hodgkin de células B, lo que coincide con la tendencia general de los linfomas oculares.
Históricamente, Arnold y Becker lo reportaron por primera vez en 1872, e Isaacson estableció el concepto de tejido linfoide asociado a mucosas (MALT) en 1984. 1) La clasificación de la OMS de 2017 define más de 80 subtipos. 1)
Q¿Cuál es la frecuencia del linfoma maligno conjuntival?
A
La incidencia es de 0,2 por cada 100.000 habitantes, y representa el 25-30% de todos los linfomas del anexo ocular. 1) Es una enfermedad rara, que corresponde al 1-2% de todos los linfomas no Hodgkin. Es más frecuente después de los 60 años y extremadamente rara en niños, con una incidencia de 0,12 por millón. 1)
Q¿Qué es el linfoma MALT?
A
El linfoma MALT (linfoma del tejido linfoide asociado a mucosas) es un término general para los linfomas de células B de bajo grado que surgen de tejidos mucosos como el tracto gastrointestinal, las glándulas salivales, los pulmones, la órbita y la conjuntiva. Se origina en el tejido linfoide adquirido localmente debido a inflamación o infección crónica, y se caracteriza por una progresión lenta y confinamiento local. Los que se originan en la conjuntiva se denominan linfoma CALT y tienen un pronóstico relativamente bueno.
McGrath LA, et al. Conjunctival Lymphoma. Eye (Lond). 2023. Figure 5. PMCID: PMC10049989. License: CC BY.
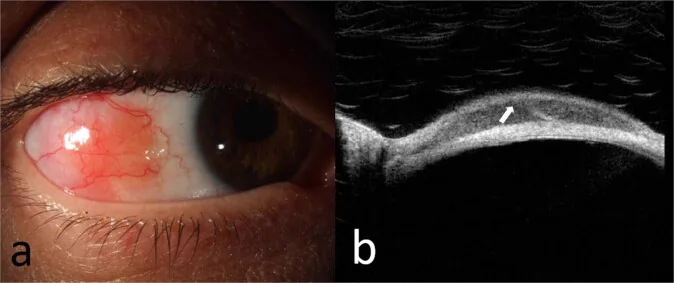
Fotografía con lámpara de hendidura (a) de un linfoma de la zona marginal extranodal de la conjuntiva bulbar, e imagen OCT (b) de la lesión que muestra una infiltración subepitelial homogénea hiporreflectiva y una banda subepitelial hiperreflectiva. Corresponde a la lesión conjuntival tratada en la sección “2. Síntomas principales y hallazgos clínicos.”
El síntoma principal es una masa conjuntival, a menudo con poca sensación de cuerpo extraño. Algunos casos se diagnostican después de un tratamiento prolongado como conjuntivitis crónica.
Masa/Protuberancia: Se percibe como una masa conjuntival que aumenta lentamente.
Sensación de cuerpo extraño/malestar leve: El 85% presenta algún síntoma, y el 67% se considera leve. 1)
Ptosis: Puede ocurrir debido al aumento de la masa.
Tiempo desde el inicio de los síntomas hasta la consulta: En casos de bajo grado, los síntomas suelen presentarse 4-6 meses antes.
Hallazgos clínicos (hallazgos confirmados por el médico durante el examen)
Aparecen lesiones elevadas, translúcidas, de color salmón en el fondo de saco y la conjuntiva bulbar. Es difícil distinguirlas de la hiperplasia linfoide reactiva (HLR) solo por su apariencia.
El sitio más común es el fondo de saco conjuntival, y a medida que progresa, puede extenderse a toda la conjuntiva. También es importante examinar la conjuntiva del párpado superior evertiéndola. Más del 90% son de color rosa, con hallazgos típicos de un diámetro basal de 15 mm, un grosor de 3 mm y 2-3 lesiones por ojo. 1) La afectación unilateral representa 2/3 de los casos (82% para el linfoma de la zona marginal extranodal), y el 62% ocurre en la conjuntiva superior o inferior. Solo el 7% alcanza el limbo. 1) Puede ocurrir afectación bilateral, ya sea simultáneamente o después de un intervalo de años en el ojo contralateral.
Las características clínicas por subtipo son las siguientes:
Linfoma de la zona marginal extranodal
Aspecto: Lesiones móviles, planas, en parches, de color salmón.
Distribución: Común en el fondo de saco y la conjuntiva bulbar. Mayoritariamente unilateral (82%). 1)
Características: Bajo grado, progresión lenta. Subtipo más común (81%). Buen pronóstico.
Linfoma folicular / de células del manto
Linfoma folicular: A menudo se presenta como lesiones multinodulares. 1)
Linfoma de células del manto: Masa grande, de color rojo oscuro. 1)
Características: El linfoma folicular es de bajo grado. El linfoma de células del manto a menudo se asocia con enfermedad sistémica.
Linfoma difuso de células B grandes / de células T
Linfoma difuso de células B grandes: Masa grisácea de crecimiento rápido. 1)
Linfoma de células T: El 30% ocurre en el limbo. Algunos casos se acompañan de escleritis. 1)
Características: Alto grado. Se requiere manejo urgente.
Q¿Masa de color salmón = linfoma?
A
Una masa conjuntival de color salmón también puede tener una apariencia similar en la hiperplasia linfoide reactiva. La diferenciación solo por apariencia es difícil, y siempre se requiere una biopsia para el diagnóstico definitivo. Consulte la sección “Diagnóstico y métodos de prueba” para más detalles.
Se cree que la patogénesis del linfoma maligno conjuntival implica una desregulación de las células B debido a la estimulación antigénica crónica. Normalmente, no hay tejido linfoide en la conjuntiva, pero la inflamación crónica persistente conduce a la adquisición de tejido linfoide asociado a mucosas, a partir del cual se origina el linfoma MALT.
Agentes infecciosos: Se han reportado asociaciones con Helicobacter pylori, Chlamydophila psittaci, virus de la hepatitis C y VIH. 1) La asociación con C. psittaci se reporta con mayor frecuencia en Europa, y la tasa de positividad varía según la región.
Enfermedades autoinmunes: Asociaciones con síndrome de Sjögren, tiroiditis de Hashimoto y enfermedad relacionada con IgG4. 1)
Microbiota conjuntival: Se ha sugerido la posible participación de Delftia sp. (bacilo gramnegativo). 1)
Asociación con hiperplasia linfoide reactiva: La hiperplasia linfoide reactiva se considera una condición precursora. 1)
El examen histopatológico mediante biopsia es esencial para el diagnóstico definitivo. No se puede diagnosticar solo con hallazgos clínicos o imágenes. El linfoma maligno es monoclonal, mientras que la hiperplasia linfoide reactiva es policlonal, y este punto es clave para la diferenciación.
La biopsia implica la extirpación parcial o completa del tumor para su evaluación patológica. Se recomienda obtener al menos 250 mg de muestra de biopsia (necesario para las pruebas de reordenamiento génico). Dado que el linfoma conjuntival crece de manera delgada y extensa, pueden ser necesarias técnicas como dividir la escisión en dos sitios para evitar la simbléfaron.
Tinción HE + inmunohistoquímica: Confirmar el linaje y la clonalidad de la población linfocítica. Utilizar CD10, CD20, CD79a, CD5, etc.
Citometría de flujo: Se puede realizar si hay suficiente muestra. Permite determinar rápidamente el linaje y el grado de diferenciación de las células tumorales.
Análisis de reordenamiento génico: Examinar la presencia de reordenamientos de los genes del receptor de inmunoglobulina/T mediante Southern blot o PCR. En el linfoma maligno, un patrón de reordenamiento monoclonal es consistente en todas las células tumorales, distinguiéndolo de la hiperplasia linfoide reactiva policlonal.
Los perfiles inmunológicos por subtipo son los siguientes:
Una vez diagnosticado un linfoma maligno, se debe realizar una búsqueda sistémica del tumor primario. Es importante colaborar con un hematólogo para la estadificación.
Para la evaluación sistémica se utilizan los siguientes:
FDG-PET/TC: Se utiliza para la estadificación sistémica. 1)
Biopsia de médula ósea: Evalúa la presencia de diseminación sistémica.
LDH sérica y β2-microglobulina: Se utilizan como marcadores tumorales de referencia.
La estadificación se evalúa mediante la clasificación de Ann Arbor (la clasificación de Lugano también puede aplicarse para el linfoma de los anexos oculares) y determina directamente la estrategia de tratamiento.
Estadio
Definición
Pauta de tratamiento
Estadio IE (localizado)
Confinado a la conjuntiva o anexos oculares
Radioterapia como primera opción
Estadio IIE o superior
Afectación de ganglios linfáticos u otros órganos
Considerar quimioterapia sistémica
Pruebas de imagen auxiliares y diagnóstico diferencial
Tomografía de coherencia óptica de alta resolución (HR-OCT): Puede usarse para evaluación complementaria, pero el diagnóstico definitivo solo es posible mediante patología. 1)
Q¿Se puede diagnosticar el linfoma conjuntival sin biopsia?
A
La tomografía de coherencia óptica de alta resolución proporciona información complementaria, pero el diagnóstico definitivo solo es posible mediante el examen histopatológico de una biopsia. 1) La PET/TC es útil para la estadificación sistémica, pero no se utiliza para el diagnóstico local. La confirmación de monoclonalidad mediante análisis de reordenamiento génico (PCR o Southern blot) constituye la base del diagnóstico definitivo.
El tratamiento se realiza en colaboración entre hematología y oftalmología. La resección quirúrgica completa es difícil; la enfermedad es altamente radiosensible y también responde bien a la quimioterapia. La estrategia de tratamiento se determina según el estadio (clasificación de Ann Arbor) y el subtipo histológico.
Cuando se sospecha hiperplasia linfoide reactiva, se puede considerar primero la observación con gotas oftálmicas de ciclosporina A al 0.05% (2-4 veces al día) o gotas de esteroides de baja concentración. En algunos casos se puede lograr la regresión, pero si se diagnostica malignidad, se debe pasar rápidamente a los siguientes tratamientos.
Tratamiento de la enfermedad localizada (estadio IE de Ann Arbor)
La radioterapia es la primera opción. Es particularmente efectiva para el linfoma de la zona marginal extranodal (linfoma MALT). Durante la irradiación, el ojo se protege con un lente de contacto de plomo. También se puede usar irradiación con haz de electrones.
La irradiación externa de 24 Gy en 12 fracciones ha reportado una tasa de control local a 5 años del 89-100%. 1) La irradiación de dosis ultra baja (4 Gy en 2 fracciones) para reducir efectos secundarios ha logrado una tasa de respuesta completa del 85% y una tasa de control a 2 años del 75%. 1)
La monoterapia con rituximab (375 mg/m² × 4 ciclos) también es una opción. 1) La inyección intralesional de rituximab (50 mg) se usa en casos recurrentes o que desean tratamiento local, con una tasa de respuesta completa reportada del 73%. 1)
Para el linfoma CD20 positivo, se selecciona la terapia R-CHOP (rituximab 375 mg/m² + CHOP). El interferón alfa-2b (1-1.5 MIU por vía subcutánea tres veces por semana) ha reportado una tasa de supervivencia libre de progresión a 5 años del 85% en casos localizados de bajo grado. 1)
En casos positivos para Chlamydophila psittaci por PCR, se ha intentado la terapia antibiótica con doxiciclina 100 mg dos veces al día, con una tasa de supervivencia libre de progresión a 5 años reportada del 55%. 1)
Los datos de respuesta de cada tratamiento se muestran a continuación.
Tratamiento
Indicaciones principales
Tasa de respuesta principal
Radioterapia externa 24 Gy/12 fracciones
Primera elección para casos localizados
Control a 5 años 89–100%1)
Radioterapia externa de dosis ultrabaja 4 Gy/2 fracciones
El ensayo IELSG-19 mostró una supervivencia libre de progresión a 5 años del 68% vs 51% y una tasa de respuesta del 95% vs 86% para rituximab más clorambucilo frente a clorambucilo solo.1) Se ha informado que la crioterapia logra la regresión de las lesiones en el 98% de 42 casos.1)
Para el linfoma linfoblástico de células T, se ha informado que la terapia con hyper-CVAD+HD-MA logra una respuesta completa del 91%, una supervivencia libre de progresión a 3 años del 66% y una supervivencia global a 3 años del 70%.2)
Q¿Cuánto tiempo es necesario el seguimiento después del tratamiento?
A
Se ha informado que el linfoma de zona marginal extranodal progresa a enfermedad sistémica en aproximadamente el 20% de los casos incluso después del tratamiento, y ocasionalmente se observa diseminación después de más de 10 años.1) Se necesitan controles regulares a largo plazo para la detección temprana de recurrencia y progresión sistémica. El intervalo y la duración del seguimiento deben determinarse en consulta con el médico tratante.
El linfoma maligno conjuntival se origina en el tejido linfoide asociado a la conjuntiva (presente en el limbo y el fondo de saco). Normalmente, no hay tejido linfoide en la conjuntiva, pero la inflamación crónica, la infección o la estimulación autoinmune conducen a la adquisición de tejido linfoide asociado a mucosas, en el que se produce la linfomagénesis.
Mecanismos moleculares del linfoma de zona marginal extranodal (linfoma MALT)
El linfoma MALT surge de las células B de la zona marginal que muestran proliferación neoplásica a través de la activación crónica de la vía NF-κB.1)
Inactivación del gen A20: Se encuentra en aproximadamente el 30% de los casos, desregula la vía NF-κB y es un factor de mal pronóstico. 1)
Mutación de MYD88: Se encuentra en aproximadamente el 7% de los casos y se asocia con mal pronóstico. 1)
Translocaciones cromosómicas: Se han reportado t(14;18) 0–38%, t(11;18) 0–15%, t(3;14) 0–14%. 1)
Anomalías en el número de copias: Se observan trisomías 3, 12 y 18, y las anomalías del cromosoma 6 se consideran específicas de los anexos oculares. 1)
El análisis de los reordenamientos de los genes del receptor de inmunoglobulina/células T es útil para determinar si una proliferación es neoplásica (maligna) o reactiva (benigna). Si se confirma una proliferación monoclonal, se considera maligna; si es policlonal, se trata como reactiva. Incluso si los hallazgos clínicos son similares, esta prueba de biología molecular es esencial para un diagnóstico definitivo.
Sugawara et al. (2022) reportaron un caso extremadamente raro de linfoma linfoblástico de células T precursoras que surgió en la conjuntiva. Un hombre de 61 años presentó una masa conjuntival de color salmón, que era CD7+, CD10+, TdT+ y CD20−. La tasa de supervivencia global a 5 años para el linfoma linfoblástico de células T en adultos, que representa aproximadamente el 2% de todos los linfomas, es pobre, del 26%. 2)
7. Investigación más reciente y perspectivas futuras
El análisis exhaustivo de mutaciones genéticas mediante secuenciación de próxima generación está avanzando en la identificación de marcadores pronósticos. 1) La expresión de BCL-6, MUM1/IRF4 y Ki-67, un diámetro tumoral >30 mm y Ki-67 >10% se destacan como factores de mal pronóstico. 1)
Monitorización del tratamiento con tomografía de coherencia óptica de alta resolución
Se está investigando la utilidad de la monitorización no invasiva del efecto del tratamiento mediante tomografía de coherencia óptica de alta resolución. 1) Podría permitir el seguimiento de los cambios tumorales sin repetir biopsias.
Se ha sugerido una asociación entre la microbiota conjuntival, incluyendo Delftia sp. (bacilos gramnegativos), y el desarrollo de linfoma conjuntival, y se está investigando como una nueva vía de estimulación antigénica crónica. 1)
Se está investigando la aplicación de fármacos de terapia molecular dirigida, incluidos los inhibidores de BTK, al linfoma conjuntival. 1) Se esperan como nuevas opciones de tratamiento para casos recidivantes/refractarios.
McGrath LA, Ryan DA, Warrier SK, Coupland SE, Glasson WJ. Conjunctival Lymphoma. Eye. 2023;37:837-848.
Sugawara R, Usui Y, Takahashi R, Nagao T, Goto H.. A case of conjunctival precursor T cell lymphoblastic lymphoma presenting with salmon colored conjunctival mass. Am J Ophthalmol Case Rep. 2022;25:101382. doi:10.1016/j.ajoc.2022.101382. PMID:35243143; PMCID:PMC8859792.
Baltă AC, Mihai MA, Ionescu AM, Radu M, Chițac I, Murgoi G, et al. Conjunctival lymphoma: case report. Rom J Ophthalmol. 2025;69(3):440-449. PMID: 41189780.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.