Das maligne Lymphom der Bindehaut ist ein primäres malignes Lymphom der Bindehaut, das durch monoklonale Proliferation von B-Zellen entsteht. Es geht vom bindehautassoziierten lymphatischen Gewebe (CALT) aus und macht 25–30 % der Lymphome der Augenanhangsgebilde aus. 1)
Die Mehrheit davon sind MALT-Lymphome (Lymphome des mukosaassoziierten lymphatischen Gewebes). Das primäre MALT-Lymphom der Bindehaut wird auch als CALT-Lymphom bezeichnet und zeichnet sich durch niedrige Malignität, langsames Fortschreiten und Neigung zur Schleimhautlokalisation aus, was es in Bezug auf Natur und Prognose deutlich von anderen hochmalignen Lymphomen unterscheidet. Diffuse mittelzellige und großzellige Lymphome hingegen weisen viele Mitosefiguren auf und sind wahrscheinlicher Metastasen von Lymphomen anderer Organe.
Die Inzidenz ist selten mit 0,2 Fällen pro 100.000 Personen, was 1–2 % aller Non-Hodgkin-Lymphome entspricht. 1) Sie tritt hauptsächlich nach dem 60. Lebensjahr auf, und die Inzidenz des extranodalen Marginalzonen-Lymphoms und des follikulären Lymphoms zeigte zwischen 1980 und 2005 einen steigenden Trend. 1) Obwohl sie häufiger bei älteren Menschen auftritt, wurden auch Fälle im Alter von 33 Monaten berichtet. Die Inzidenz von Lymphomen der Augenanhangsgebilde bei Kindern ist mit 0,12 Fällen pro Million Einwohner extrem selten. 1)
Die histologischen Subtypen sind in einer Zusammenstellung von 1014 Fällen: extranodales Marginalzonen-Lymphom 81 %, follikuläres Lymphom (FL) 8 %, Mantelzell-Lymphom (MCL) 3 %, diffuses großzelliges B-Zell-Lymphom (DLBCL) 3 %, und 98 % sind vom B-Zell-Typ. 1) Die meisten sind Non-Hodgkin-B-Zell-Lymphome, was dem allgemeinen Trend der Lymphome im Augenbereich entspricht.
Historisch gesehen wurde der erste Fall 1872 von Arnold und Becker berichtet, und 1984 etablierte Isaacson das Konzept des mukosaassoziierten lymphatischen Gewebes (MALT). 1) Die WHO-Klassifikation von 2017 definiert über 80 Subtypen. 1)
QWie häufig ist das maligne Lymphom der Konjunktiva?
A
Die Inzidenz beträgt 0,2 Fälle pro 100.000 Personen und macht 25–30 % aller Lymphome der Augenanhangsgebilde aus. 1) Es ist eine seltene Erkrankung, die 1–2 % aller Non-Hodgkin-Lymphome ausmacht. Sie tritt hauptsächlich nach dem 60. Lebensjahr auf und ist bei Kindern mit 0,12 Fällen pro Million Kinder äußerst selten. 1)
QWas ist ein MALT-Lymphom?
A
Das MALT-Lymphom (Mukosa-assoziiertes lymphatisches Gewebe-Lymphom) ist ein Sammelbegriff für niedrigmaligne B-Zell-Lymphome, die aus Schleimhautgeweben wie dem Gastrointestinaltrakt, Speicheldrüsen, Lunge, Orbita und Konjunktiva entstehen. Es entsteht aus lokal erworbenem lymphatischem Gewebe aufgrund chronischer Entzündung oder Infektion und zeichnet sich durch langsames Fortschreiten und lokale Begrenzung aus. Die aus der Konjunktiva stammenden werden als CALT-Lymphome bezeichnet und haben eine relativ gute Prognose.
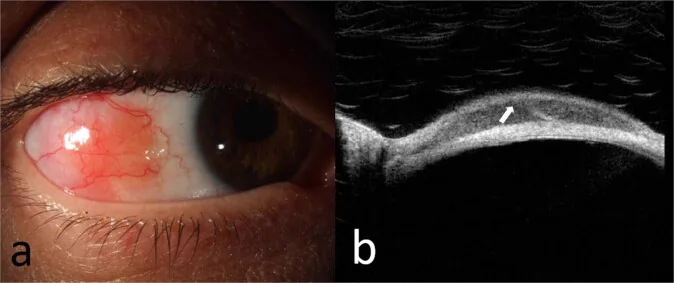
McGrath LA, et al. Conjunctival Lymphoma. Eye (Lond). 2023. Figure 5. PMCID: PMC10049989. License: CC BY.
Spaltlampenfotografie (a) eines extranodalen Marginalzonen-Lymphoms der bulbären Konjunktiva und OCT (b) der Läsion, die ein hyporeflektives, homogenes subepitheliales Infiltrat und ein hyperreflektives subepitheliales Band zeigt. Dies entspricht den konjunktivalen Läsionen, die im Abschnitt „2. Hauptsymptome und klinische Befunde“ behandelt werden.
Das Hauptsymptom ist eine konjunktivale Raumforderung, die oft nur wenig Fremdkörpergefühl verursacht. In einigen Fällen wird die Diagnose nach langer Behandlung als chronische Konjunktivitis gestellt.
Raumforderung/Vorwölbung: wird als langsam wachsende konjunktivale Raumforderung wahrgenommen.
Fremdkörpergefühl/leichtes Unbehagen: 85 % haben Symptome, davon 67 % leichte. 1)
Ptosis: kann durch die Vergrößerung der Raumforderung auftreten.
Zeit vom Symptombeginn bis zur Konsultation: Bei niedrigmalignen Formen bestehen die Symptome oft seit 4–6 Monaten.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Lachsfarbene, durchscheinende, erhabene Läsionen treten am Fornix und an der Bulbärkonjunktiva auf. Eine visuelle Unterscheidung von einer reaktiven lymphoiden Hyperplasie (RLH) ist schwierig.
Die bevorzugte Lokalisation ist die Fornixkonjunktiva, und bei Fortschreiten kann sie sich auf die gesamte Konjunktiva ausdehnen. Es ist auch wichtig, die Konjunktiva des oberen Augenlids durch Umdrehen zu untersuchen. Über 90 % sind rosa gefärbt, mit einem typischen Basisdurchmesser von 15 mm, einer Dicke von 3 mm und 2–3 Läsionen pro Auge. 1) Zwei Drittel sind einseitig (extranodales Marginalzonenlymphom 82 %), 62 % treten in der oberen oder unteren Konjunktiva auf. Nur 7 % erreichen den Limbus. 1) Es kann auch beidseitig auftreten, gleichzeitig oder nach Jahren am anderen Auge.
Die klinischen Merkmale nach Subtyp sind wie folgt:
Ein lachsfarbener Bindehauttumor kann auch bei reaktiver lymphoider Hyperplasie auftreten. Eine Unterscheidung allein anhand des Aussehens ist schwierig; für die endgültige Diagnose ist eine Biopsie erforderlich. Einzelheiten finden Sie im Abschnitt „Diagnose und Untersuchungsmethoden“.
Der Entstehungsmechanismus des malignen Bindehautlymphoms wird als Fehlregulation der B-Zellen durch chronische Antigenstimulation angesehen. Normalerweise gibt es in der Bindehaut kein lymphatisches Gewebe, aber eine anhaltende chronische Entzündung kann zur Bildung von schleimhautassoziiertem lymphatischem Gewebe führen, aus dem ein MALT-Lymphom entsteht.
Infektionserreger: Assoziationen mit Helicobacter pylori, Chlamydophila psittaci, Hepatitis-C-Virus und HIV wurden berichtet. 1) Die Assoziation mit C. psittaci wird häufiger aus Europa berichtet, und die Positivitätsrate variiert regional.
Autoimmunerkrankungen: Assoziation mit Sjögren-Syndrom, Hashimoto-Thyreoiditis und IgG4-assoziierten Erkrankungen. 1)
Bindehautmikrobiom: Delftia sp. (gramnegatives Stäbchen) könnte beteiligt sein. 1)
Beziehung zur reaktiven lymphoiden Hyperplasie: Die reaktive lymphoide Hyperplasie wird als Vorläuferzustand angesehen. 1)
Für die endgültige Diagnose ist eine histopathologische Untersuchung durch Biopsie zwingend erforderlich. Die Diagnose kann nicht allein aufgrund klinischer Befunde und Bildgebung gestellt werden. Das maligne Lymphom ist monoklonal, während die reaktive lymphoide Hyperplasie polyklonal ist – dies ist der Kernpunkt der Unterscheidung.
Bei der Biopsie wird ein Teil oder der gesamte Tumor entfernt und pathologisch untersucht. Es wird empfohlen, eine Biopsiemenge von mindestens 250 mg sicherzustellen (erforderlich für Gen-Rearrangement-Tests). Da das Bindehautlymphom dünn und flächig wächst, kann es erforderlich sein, die Resektionsfläche auf zwei Stellen aufzuteilen, um eine Symblepharonbildung zu vermeiden.
HE-Färbung + Immunhistochemie : Bestätigung der lymphozytären Linienzugehörigkeit und Klonalität. Verwendung von CD10, CD20, CD79a, CD5 usw.
Durchflusszytometrie : Durchführbar, wenn ausreichend Probenmaterial vorhanden ist. Ermöglicht die schnelle Bestimmung der Linie und des Differenzierungsgrades der Tumorzellen.
Gen-Rearrangement-Analyse : Mittels Southern-Blot oder PCR wird das Vorhandensein von Immunglobulin/T-Zell-Rezeptor-Gen-Rearrangements untersucht. Bei malignen Lymphomen zeigt sich ein monoklonales Rearrangement-Muster, das in allen Tumorzellen identisch ist und es von der polyklonalen reaktiven lymphatischen Hyperplasie unterscheidet.
Die immunologischen Profile nach Subtyp sind wie folgt:
Nach der Diagnose eines malignen Lymphoms wird eine systemische Suche nach einem Primärtumor durchgeführt. Es ist wichtig, die Stadieneinteilung in Zusammenarbeit mit einem Hämatologen vorzunehmen.
Die systemische Suche umfasst Folgendes:
FDG-PET/CT : Wird für die systemische Stadieneinteilung verwendet. 1)
Knochenmarkbiopsie : Beurteilt das Vorhandensein einer systemischen Dissemination.
Serum-LDH und β2-Mikroglobulin : Werden als Tumormarker zur Referenz herangezogen.
Das Stadium wird nach der Ann-Arbor-Klassifikation (bei okulären Adnexlymphomen kann auch die Lugano-Klassifikation angewendet werden) bewertet und bestimmt direkt die Therapiestrategie.
Stadium
Definition
Therapiehinweis
Stadium IE (lokalisiert)
Auf Konjunktiva oder okuläre Adnexe beschränkt
Strahlentherapie als erste Wahl
Stadium IIE oder höher
Befall von Lymphknoten oder anderen Organen
Systemische Chemotherapie in Betracht ziehen
Hilfsbildgebende Untersuchungen und Differentialdiagnose
Hochauflösende optische Kohärenztomographie (HR-OCT): Kann zur ergänzenden Beurteilung verwendet werden, aber die endgültige Diagnose erfolgt nur durch Pathologie. 1)
QKann ein Konjunktival-Lymphom ohne Biopsie diagnostiziert werden?
A
Die hochauflösende optische Kohärenztomographie liefert ergänzende Informationen, aber die endgültige Diagnose ist nur durch die histopathologische Untersuchung einer Biopsie möglich. 1) PET/CT ist für das systemische Staging nützlich, wird aber nicht für die lokale Diagnose verwendet. Der Nachweis der Monoklonalität durch Gen-Rearrangement-Analyse (PCR oder Southern Blot) ist die Grundlage für die endgültige Diagnose.
Die Behandlung erfolgt in Zusammenarbeit zwischen Hämatologie und Augenheilkunde. Eine vollständige chirurgische Resektion ist schwierig; der Tumor ist strahlenempfindlich und spricht gut auf Chemotherapie an. Die Behandlungsstrategie wird nach Stadium (Ann-Arbor-Klassifikation) und histologischem Subtyp festgelegt.
Bei Verdacht auf reaktive lymphoide Hyperplasie kann zunächst eine Beobachtung unter 0,05% Cyclosporin A Augentropfen (2-4 mal täglich) oder niedrig konzentrierten Steroid-Augentropfen erfolgen. Manchmal kommt es zu einer Rückbildung, aber bei Diagnose einer Malignität wird schnell auf die folgende Behandlung umgestellt.
Die Strahlentherapie ist die erste Wahl. Sie ist besonders wirksam bei MALT-Lymphom. Während der Bestrahlung wird das Auge durch eine Blei-Kontaktlinse geschützt. Auch eine Elektronenbestrahlung kann eingesetzt werden.
Eine externe Bestrahlung mit 24 Gy in 12 Fraktionen ergibt eine 5-Jahres-Lokalkontrollrate von 89-100 %. 1) Bei Verwendung einer ultraniedrigen Dosis (4 Gy in 2 Fraktionen) zur Reduzierung von Nebenwirkungen wurden eine vollständige Ansprechrate von 85 % und eine 2-Jahres-Kontrollrate von 75 % erzielt. 1)
Eine Rituximab-Monotherapie (375 mg/m² × 4 Zyklen) ist ebenfalls eine Option. 1) Die intraläsionale lokale Injektion von Rituximab (50 mg) wird bei Rezidiven oder bei Patienten, die eine lokale Behandlung wünschen, eingesetzt, mit einer berichteten vollständigen Ansprechrate von 73 %. 1)
Behandlung von Fällen mit systemischer Beteiligung
Bei CD20-positiven Lymphomen wird eine R-CHOP-Therapie (Rituximab 375 mg/m² + CHOP) gewählt. Interferon alpha-2b (1-1,5 Mio. IE subkutan 3-mal wöchentlich) zeigte bei niedrigmalignen, lokalisierten Fällen eine 5-Jahres-Progressionsfrei-Überlebensrate von 85 %. 1)
Bei Chlamydophila psittaci PCR-positiven Fällen wurde eine Antibiotikatherapie mit Doxycyclin 100 mg (2-mal täglich) versucht, mit einer berichteten 5-Jahres-Progressionsfrei-Überlebensrate von 55 %. 1)
Die Wirksamkeitsdaten der einzelnen Behandlungen sind unten aufgeführt.
Die IELSG-19-Studie zeigte ein 5-Jahres-progressionsfreies Überleben von 68 % vs. 51 % und eine Ansprechrate von 95 % vs. 86 % für die Gruppe mit Rituximab + Chlorambucil im Vergleich zur Gruppe mit Chlorambucil allein.1) Bei Kryotherapie wurde bei 98 % von 42 Fällen eine Rückbildung der Läsionen berichtet.1)
Für das T-Zell-lymphoblastische Lymphom wurde unter hyper-CVAD + HD-MA-Therapie eine vollständige Remission von 91 %, ein 3-Jahres-progressionsfreies Überleben von 66 % und ein 3-Jahres-Gesamtüberleben von 70 % berichtet.2)
QWie lange ist eine Nachsorge nach der Behandlung erforderlich?
A
Beim extranodalen Marginalzonenlymphom wird nach der Behandlung in etwa 20 % der Fälle ein Fortschreiten zu einer systemischen Erkrankung berichtet, und es gibt vereinzelte Fälle einer Dissemination nach mehr als 10 Jahren.1) Zur Früherkennung eines Rezidivs oder einer systemischen Progression ist eine langfristige regelmäßige Nachsorge erforderlich. Die Häufigkeit und Dauer der Nachsorge sollte mit dem behandelnden Arzt festgelegt werden.
6. Pathophysiologie und detaillierte Krankheitsmechanismen
Das maligne Konjunktivalymphom entsteht im konjunktivalen assoziierten lymphatischen Gewebe (am Limbus und Fornix). Normalerweise enthält die Konjunktiva kein lymphatisches Gewebe, aber chronische Entzündung, Infektion oder autoimmune Stimulation können zum Erwerb von mukosaassoziiertem lymphatischem Gewebe führen, in dem dann eine Lymphomentwicklung stattfindet.
Molekulare Mechanismen des extranodalen Marginalzonenlymphoms (MALT-Lymphom)
Zur Beurteilung, ob eine Proliferation neoplastisch (maligne) oder reaktiv (benigne) ist, ist die Analyse der Immunglobulin-/T-Zell-Rezeptor-Gen-Rearrangements nützlich. Wird eine monoklonale Proliferation bestätigt, gilt sie als maligne; bei polyklonaler Proliferation wird sie als reaktiv behandelt. Auch wenn die klinischen Befunde ähnlich sind, ist dieser molekularbiologische Test für die endgültige Diagnose unerlässlich.
Sugawara et al. (2022) berichteten über einen äußerst seltenen Fall eines Vorläufer-T-Zell-lymphoblastischen Lymphoms der Konjunktiva. Ein 61-jähriger Mann präsentierte sich mit einer lachsfarbenen konjunktivalen Raumforderung, die CD7+, CD10+, TdT+, CD20− zeigte. Die 5-Jahres-Gesamtüberlebensrate bei Erwachsenen mit T-Zell-lymphoblastischem Lymphom, das etwa 2 % aller Lymphome ausmacht, beträgt 26 % und ist damit schlecht. 2)
Durch umfassende Genmutationsanalyse mittels Next-Generation-Sequencing werden prognostische Marker identifiziert. 1) Die Expression von BCL-6, MUM1/IRF4 und Ki-67, Tumorgröße >30 mm und Ki-67 >10 % gelten als ungünstige Prognosefaktoren. 1)
Der Nutzen der nicht-invasiven Therapieüberwachung mittels hochauflösender optischer Kohärenztomographie wird untersucht. 1) Dies könnte eine Verfolgung von Tumorveränderungen ohne wiederholte Biopsien ermöglichen.
Konjunktivales Mikrobiom und Pathogenesemechanismus
Ein Zusammenhang zwischen dem konjunktivalen Mikrobiom, einschließlich Delftia sp. (gramnegatives Stäbchen), und der Entstehung eines konjunktivalen Lymphoms wurde vorgeschlagen, und die Forschung zu diesem neuen Weg der chronischen Antigenstimulation läuft. 1)
Die Anwendung molekular zielgerichteter Medikamente, einschließlich BTK-Inhibitoren, beim konjunktivalen Lymphom wird untersucht. 1) Sie gelten als neue Behandlungsoption für rezidivierende oder refraktäre Fälle.
McGrath LA, Ryan DA, Warrier SK, Coupland SE, Glasson WJ. Conjunctival Lymphoma. Eye. 2023;37:837-848.
Sugawara R, Usui Y, Takahashi R, Nagao T, Goto H.. A case of conjunctival precursor T cell lymphoblastic lymphoma presenting with salmon colored conjunctival mass. Am J Ophthalmol Case Rep. 2022;25:101382. doi:10.1016/j.ajoc.2022.101382. PMID:35243143; PMCID:PMC8859792.
Baltă AC, Mihai MA, Ionescu AM, Radu M, Chițac I, Murgoi G, et al. Conjunctival lymphoma: case report. Rom J Ophthalmol. 2025;69(3):440-449. PMID: 41189780.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.