Le lymphome malin conjonctival est un lymphome malin primitif de la conjonctive dû à une prolifération monoclonale de cellules B. Il provient du tissu lymphoïde associé à la conjonctive (CALT) et représente 25 à 30 % des lymphomes des annexes oculaires. 1)
La majorité d’entre eux correspond à un lymphome MALT (lymphome du tissu lymphoïde associé aux muqueuses). Le lymphome MALT primitif de la conjonctive est également appelé lymphome du CALT, avec une tendance à la faible malignité, à la progression lente et à la localisation muqueuse, ce qui le distingue nettement des autres lymphomes de haute malignité en termes de nature et de pronostic. En revanche, les lymphomes à cellules de taille moyenne ou grande présentent de nombreuses figures mitotiques et sont plus susceptibles d’être des métastases d’un lymphome d’autres organes.
L’incidence est rare, avec 0,2 cas pour 100 000 personnes, correspondant à 1 à 2 % de tous les lymphomes non hodgkiniens. 1) Elle survient principalement après 60 ans, et l’incidence du lymphome de la zone marginale extranodal et du lymphome folliculaire a augmenté entre 1980 et 2005. 1) Bien que plus fréquent chez les personnes âgées, des cas ont été rapportés dès l’âge de 33 mois. L’incidence du lymphome des annexes oculaires chez l’enfant est extrêmement rare, avec 0,12 cas par million d’habitants. 1)
Les sous-types histologiques, selon une compilation de 1014 cas, sont : lymphome de la zone marginale extraganglionnaire 81 %, lymphome folliculaire (FL) 8 %, lymphome à cellules du manteau (MCL) 3 %, lymphome diffus à grandes cellules B (DLBCL) 3 %, et 98 % sont de type B. 1) La plupart sont des lymphomes B non hodgkiniens, ce qui correspond à la tendance générale des lymphomes ophtalmologiques.
Historiquement, Arnold et Becker ont rapporté le premier cas en 1872, et Isaacson a établi le concept de tissu lymphoïde associé aux muqueuses (MALT) en 1984. 1) La classification OMS 2017 définit plus de 80 sous-types. 1)
QQuelle est la fréquence du lymphome malin conjonctival ?
A
L’incidence est de 0,2 cas pour 100 000 personnes, représentant 25 à 30 % de tous les lymphomes des annexes oculaires. 1) C’est une maladie rare, correspondant à 1 à 2 % de tous les lymphomes non hodgkiniens. Elle survient principalement après 60 ans, et est extrêmement rare chez l’enfant, avec 0,12 cas par million d’enfants. 1)
QQu'est-ce que le lymphome MALT ?
A
Le lymphome MALT (lymphome du tissu lymphoïde associé aux muqueuses) est un terme générique désignant les lymphomes B de bas grade provenant des tissus muqueux tels que le tube digestif, les glandes salivaires, les poumons, l’orbite et la conjonctive. Il provient du tissu lymphoïde acquis localement en raison d’une inflammation chronique ou d’une infection, et se caractérise par une progression lente et une localisation locale. Ceux provenant de la conjonctive sont appelés lymphomes CALT et ont un pronostic relativement favorable.
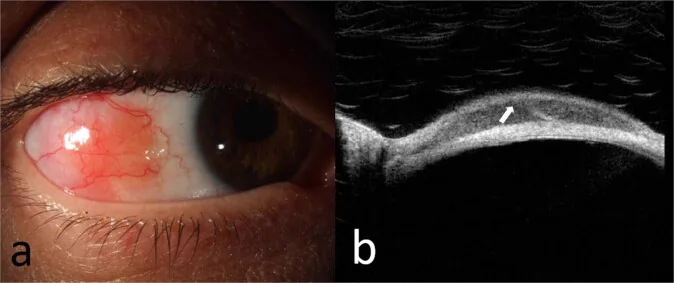
McGrath LA, et al. Conjunctival Lymphoma. Eye (Lond). 2023. Figure 5. PMCID: PMC10049989. License: CC BY.
Photographie à la lampe à fente (a) d’un lymphome de la zone marginale extraganglionnaire de la conjonctive bulbaire et image OCT (b) de la lésion montrant un infiltrat sous-épithélial homogène hypo-réfléchissant et une bande sous-épithéliale hyper-réfléchissante. Cela correspond aux lésions conjonctivales traitées dans la section « 2. Principaux symptômes et signes cliniques ».
Le principal symptôme est une masse conjonctivale, souvent avec peu de sensation de corps étranger. Certains cas sont diagnostiqués après un long traitement comme conjonctivite chronique.
Masse/saillie : perçue comme une masse conjonctivale qui augmente lentement.
Sensation de corps étranger/léger inconfort : 85 % présentent des symptômes, dont 67 % légers. 1)
Ptosis : peut survenir en raison de l’augmentation de la masse.
Délai entre l’apparition des symptômes et la consultation : dans les lymphomes de bas grade, les symptômes sont souvent présents depuis 4 à 6 mois.
Signes cliniques (constatations observées par le médecin lors de l’examen)
Des lésions surélevées, translucides, de couleur saumon apparaissent sur la conjonctive bulbaire et au niveau du fornix. Il est difficile de les distinguer visuellement d’une hyperplasie lymphoïde réactionnelle (RLH).
Le site de prédilection est la conjonctive du fornix, et en progressant, la lésion peut s’étendre à toute la conjonctive. Il est important d’examiner la conjonctive palpébrale supérieure en la retournant. Dans plus de 90 % des cas, la lésion est de couleur rose, avec un diamètre de base typique de 15 mm, une épaisseur de 3 mm et 2 à 3 lésions par œil. 1) La maladie est unilatérale dans 2/3 des cas (lymphome de la zone marginale extraganglionnaire dans 82 %), et 62 % des lésions se situent dans la conjonctive supérieure ou inférieure. Seulement 7 % atteignent le limbe. 1) La maladie peut également être bilatérale, survenant simultanément ou après un intervalle de plusieurs années dans l’œil controlatéral.
Les caractéristiques cliniques selon le sous-type sont les suivantes :
Lymphome de la zone marginale extraganglionnaire
Aspect : lésion maculaire plate, mobile, de couleur saumon.
Distribution : prédominance au fornix et à la conjonctive bulbaire. Souvent unilatéral (82 %). 1)
Caractéristiques : de bas grade, progression lente. Sous-type le plus fréquent (81 %). Bon pronostic.
Lymphome folliculaire / lymphome à cellules du manteau
Lymphome folliculaire : se présente souvent sous forme de lésions multinodulaires. 1)
Lymphome à cellules du manteau : masse volumineuse, rouge foncé. 1)
Caractéristiques : le lymphome folliculaire est de bas grade. Le lymphome à cellules du manteau est souvent associé à une atteinte systémique.
Lymphome B diffus à grandes cellules / lymphome T
Lymphome B diffus à grandes cellules : masse grisâtre à croissance rapide. 1)
Lymphome T : 30 % surviennent au niveau du limbe. Certains cas s’accompagnent de sclérite. 1)
Caractéristiques : Haut grade. Une prise en charge urgente est nécessaire.
QMasse rose saumon = lymphome ?
A
Une masse conjonctivale rose saumon peut également apparaître dans l’hyperplasie lymphoïde réactive. La différenciation sur le seul aspect est difficile, et une biopsie est indispensable pour le diagnostic définitif. Voir la section « Diagnostic et méthodes d’examen » pour plus de détails.
Le mécanisme de développement du lymphome malin conjonctival est considéré comme une dérégulation des cellules B due à une stimulation antigénique chronique. Normalement, il n’y a pas de tissu lymphoïde dans la conjonctive, mais une inflammation chronique persistante peut conduire à l’acquisition d’un tissu lymphoïde associé aux muqueuses, à partir duquel un lymphome MALT se développe.
Agents infectieux : Des associations avec Helicobacter pylori, Chlamydophila psittaci, le virus de l’hépatite C et le VIH ont été rapportées. 1) L’association avec C. psittaci est fréquemment rapportée en Europe, et le taux de positivité varie selon les régions.
Maladies auto-immunes : Association avec le syndrome de Sjögren, la thyroïdite de Hashimoto et les maladies liées aux IgG4. 1)
Microbiote conjonctival : Delftia sp. (bacille à Gram négatif) pourrait être impliqué. 1)
Relation avec l’hyperplasie lymphoïde réactive : L’hyperplasie lymphoïde réactive est considérée comme un état précurseur. 1)
Un examen histopathologique par biopsie est indispensable pour le diagnostic définitif. Le diagnostic ne peut être posé sur la seule base des signes cliniques et de l’imagerie. Le lymphome malin est monoclonal, tandis que l’hyperplasie lymphoïde réactive est polyclonale, ce qui constitue le point central de la différenciation.
La biopsie consiste à prélever une partie ou la totalité de la tumeur pour un examen pathologique. Il est recommandé d’obtenir au moins 250 mg de tissu (nécessaire pour les tests de réarrangement génique). Comme le lymphome conjonctival se développe en une fine couche étendue, il peut être nécessaire de diviser la zone de résection en deux sites pour éviter une symblépharon.
Coloration HE + immunohistochimie : confirmer la lignée lymphocytaire et la clonalité. Utiliser CD10, CD20, CD79a, CD5, etc.
Cytométrie en flux : réalisable si la quantité d’échantillon est suffisante. Permet de déterminer rapidement la lignée et le degré de différenciation des cellules tumorales.
Analyse de réarrangement génique : par Southern blot ou PCR pour détecter la présence de réarrangements des gènes des immunoglobulines/récepteurs des cellules T. Dans le lymphome malin, un motif de réarrangement monoclonal est identique dans toutes les cellules tumorales, ce qui le distingue de l’hyperplasie lymphoïde réactionnelle polyclonale.
Les profils immunologiques par sous-type sont les suivants :
Sous-type
Marqueurs caractéristiques
Lymphome de la zone marginale extraganglionnaire (EMZL / MALT)
Une fois le diagnostic de lymphome malin posé, une recherche systémique est effectuée pour détecter une éventuelle lésion primitive. Il est important de réaliser un bilan de stade en collaboration avec un hématologue.
La recherche systémique comprend les examens suivants :
FDG-PET/CT : utilisé pour la classification par stade systémique. 1)
Biopsie de la moelle osseuse : évalue la présence d’une dissémination systémique.
LDH sérique et β2-microglobuline : utilisés comme marqueurs tumoraux de référence.
Le stade est évalué selon la classification d’Ann Arbor (la classification de Lugano peut également être appliquée pour le lymphome des annexes oculaires) et détermine directement la stratégie thérapeutique.
Stade
Définition
Indication thérapeutique
Stade IE (localisé)
Limité à la conjonctive ou aux annexes oculaires
Radiothérapie en première intention
Stade IIE ou plus
Atteinte ganglionnaire ou d’autres organes
Chimiothérapie systémique à envisager
Examens d’imagerie auxiliaires et diagnostic différentiel
Tomographie par cohérence optique à haute résolution (HR-OCT) : Peut être utilisée pour une évaluation auxiliaire, mais le diagnostic définitif repose uniquement sur l’histopathologie. 1)
Diagnostics différentiels : Il est nécessaire de différencier l’hyperplasie lymphoïde réactionnelle, la maladie liée aux IgG4, la conjonctivite chronique, le ptérygion, le granulome pyogène, etc. 1)
QPeut-on diagnostiquer un lymphome conjonctival autrement que par biopsie ?
A
La tomographie par cohérence optique à haute résolution fournit des informations auxiliaires, mais le diagnostic définitif n’est possible que par l’examen histopathologique d’une biopsie. 1) La TEP/TDM est utile pour la stadification systémique, mais pas pour le diagnostic local. La confirmation de la clonalité par analyse de réarrangement génique (PCR ou Southern blot) constitue la base du diagnostic définitif.
Le traitement est effectué en collaboration entre l’hématologie et l’ophtalmologie. La résection chirurgicale complète est difficile ; la tumeur est radiosensible et répond bien à la chimiothérapie. La stratégie thérapeutique est déterminée en fonction du stade (classification d’Ann Arbor) et du sous-type histologique.
En cas de suspicion d’hyperplasie lymphoïde réactionnelle, on peut d’abord observer l’évolution sous collyre de ciclosporine A à 0,05 % (2 à 4 fois par jour) ou sous collyre de corticostéroïdes à faible concentration. Une régression peut être obtenue, mais si un diagnostic de malignité est posé, on passe rapidement au traitement suivant.
Traitement des cas localisés (stade IE d’Ann Arbor)
La radiothérapie est le traitement de première intention. Elle est particulièrement efficace dans le lymphome du tissu lymphoïde associé aux muqueuses (lymphome MALT). Pendant l’irradiation, l’œil est protégé par une lentille de contact en plomb. Une irradiation par faisceau d’électrons peut également être utilisée.
Une radiothérapie externe de 24 Gy en 12 fractions a montré un taux de contrôle local à 5 ans de 89 à 100 %. 1) L’utilisation d’une irradiation à très faible dose (4 Gy en 2 fractions) pour réduire les effets secondaires a permis d’obtenir un taux de réponse complète de 85 % et un taux de contrôle à 2 ans de 75 %. 1)
L’administration de rituximab en monothérapie (375 mg/m² × 4 cycles) est également une option. 1) L’injection locale intralésionnelle de rituximab (50 mg) est utilisée dans les cas de récidive ou chez les patients souhaitant un traitement local, avec un taux de réponse complète rapporté de 73 %. 1)
Pour les lymphomes CD20 positifs, le traitement R-CHOP (rituximab 375 mg/m² + CHOP) est choisi. L’interféron alpha-2b (1 à 1,5 MUI en injection sous-cutanée 3 fois par semaine) a montré un taux de survie sans progression à 5 ans de 85 % dans les cas de bas grade et localisés. 1)
Dans les cas positifs pour Chlamydophila psittaci par PCR, un traitement antibiotique par doxycycline 100 mg (2 fois par jour) a été essayé, avec un taux de survie sans progression à 5 ans rapporté de 55 %. 1)
Les données d’efficacité de chaque traitement sont présentées ci-dessous.
Traitement
Principales indications
Taux de réponse principal
Radiothérapie externe 24 Gy/12 fractions
Première intention pour les cas localisés
Contrôle à 5 ans 89–100 %1)
Radiothérapie externe à très faible dose 4 Gy/2 fractions
Cas souhaitant une réduction des effets secondaires
L’essai IELSG-19 a montré une survie sans progression à 5 ans de 68 % vs 51 % et un taux de réponse de 95 % vs 86 % pour le groupe rituximab + chlorambucil par rapport au groupe chlorambucil seul.1) La cryothérapie a rapporté une régression des lésions chez 98 % des 42 cas.1)
Pour le lymphome lymphoblastique à cellules T, un taux de réponse complète de 91 %, une survie sans progression à 3 ans de 66 % et une survie globale à 3 ans de 70 % ont été rapportés avec le traitement hyper-CVAD + HD-MA.2)
QCombien de temps le suivi après traitement est-il nécessaire ?
A
Le lymphome de la zone marginale extraganglionnaire évolue vers une atteinte systémique dans environ 20 % des cas après traitement, et des cas de dissémination après plus de 10 ans sont parfois observés.1) Un suivi régulier à long terme est nécessaire pour détecter précocement une récidive ou une progression systémique. La fréquence et la durée du suivi doivent être déterminées avec le médecin traitant.
6. Physiopathologie et mécanismes détaillés de la maladie
Le lymphome malin conjonctival prend naissance dans le tissu lymphoïde associé à la conjonctive (situé au limbe et au fornix). Normalement, la conjonctive ne contient pas de tissu lymphoïde, mais une inflammation chronique, une infection ou une stimulation auto-immune peut entraîner l’acquisition de tissu lymphoïde associé aux muqueuses, qui peut ensuite subir une transformation lymphomateuse.
Mécanismes moléculaires du lymphome de la zone marginale extraganglionnaire (lymphome MALT)
Pour déterminer si une prolifération est tumorale (maligne) ou réactionnelle (bénigne), l’analyse du réarrangement des gènes des immunoglobulines/récepteurs des cellules T est utile. Une prolifération monoclonale confirme la malignité, tandis qu’une prolifération polyclonale indique une réaction bénigne. Même si les présentations cliniques sont similaires, ce test de biologie moléculaire est essentiel pour le diagnostic définitif.
Sugawara et al. (2022) ont rapporté un cas extrêmement rare de lymphome lymphoblastique T précurseur survenant dans la conjonctive. Un homme de 61 ans présentait une masse conjonctivale rose saumon, avec un immunophénotype CD7+, CD10+, TdT+, CD20−. La survie globale à 5 ans chez l’adulte pour le lymphome lymphoblastique T, qui représente environ 2 % de tous les lymphomes, est faible, à 26 %. 2)
L’analyse complète des mutations génétiques par séquençage de nouvelle génération permet d’identifier des marqueurs pronostiques. 1) L’expression de BCL-6, MUM1/IRF4 et Ki-67, une taille tumorale >30 mm et un Ki-67 >10 % sont considérés comme des facteurs de mauvais pronostic. 1)
Suivi thérapeutique par tomographie par cohérence optique à haute résolution
L’utilité du suivi non invasif de l’efficacité thérapeutique par tomographie par cohérence optique à haute résolution est à l’étude. 1) Cela pourrait permettre de suivre l’évolution tumorale sans biopsies répétées.
Une association entre le microbiote conjonctival, incluant Delftia sp. (bacille à Gram négatif), et le développement du lymphome conjonctival a été suggérée, et des recherches sont en cours sur cette nouvelle voie de stimulation antigénique chronique. 1)
L’application de médicaments ciblés moléculaires, notamment les inhibiteurs de BTK, au lymphome conjonctival est à l’étude. 1) Ils sont considérés comme une nouvelle option thérapeutique pour les cas récidivants ou réfractaires.
McGrath LA, Ryan DA, Warrier SK, Coupland SE, Glasson WJ. Conjunctival Lymphoma. Eye. 2023;37:837-848.
Sugawara R, Usui Y, Takahashi R, Nagao T, Goto H.. A case of conjunctival precursor T cell lymphoblastic lymphoma presenting with salmon colored conjunctival mass. Am J Ophthalmol Case Rep. 2022;25:101382. doi:10.1016/j.ajoc.2022.101382. PMID:35243143; PMCID:PMC8859792.
Baltă AC, Mihai MA, Ionescu AM, Radu M, Chițac I, Murgoi G, et al. Conjunctival lymphoma: case report. Rom J Ophthalmol. 2025;69(3):440-449. PMID: 41189780.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.