La sclérite est une inflammation des vaisseaux profonds, notamment le plexus vasculaire épiscléral et intrascléral, accompagnée d’œdème et d’infiltration cellulaire de la sclère. La sclère est un tissu fibreux peu vascularisé, et la sclérite profonde est une maladie rare. Elle peut être unilatérale ou bilatérale, et les causes sont idiopathiques, associées à une maladie systémique, infectieuses ou postopératoires.
L’incidence est rapportée entre 1,6 et 5,5 pour 100 000 personnes-années 5). Elle est plus fréquente chez les femmes. Pour les formes non nécrosantes diffuses et nodulaires, l’âge de prédilection est la quarantaine ; pour la sclérite nécrosante, la soixantaine. Le taux d’atteinte bilatérale dans la sclérite nécrosante est d’environ 60 %. La majorité des cas sont non infectieux, souvent associés à une maladie inflammatoire systémique. La sclérite infectieuse est rare (5 à 10 %) mais de mauvais pronostic 7).
Selon une enquête multicentrique japonaise basée sur les directives cliniques pour l’uvéite, sur 3 810 patients consultant pour uvéite, 235 (6,2 %) présentaient une sclérite, ce qui en fait la deuxième cause la plus fréquente après l’uvéite antérieure aiguë (6,6 %) 8).
La classification clinique classique de Watson (Watson et al., 1976) est largement utilisée. Elle distingue la sclérite antérieure et postérieure, et subdivise la sclérite antérieure en trois types selon la morphologie.
Classification
Type
Caractéristiques
Sclérite antérieure
Diffuse
La plus fréquente. Hyperémie diffuse due à la dilatation et à la tortuosité des vaisseaux scléraux.
Sclérite antérieure
Nodulaire
Nodule scléral rouge foncé. Survient fréquemment au niveau du limbe et de la zone palpébrale
Sclérite antérieure
Nécrosante (inflammatoire)
Risque de nécrose sclérale, amincissement, perforation
La sclérite diffuse est la plus fréquente, suivie de la sclérite nodulaire. La sclérite nécrosante et la sclérite postérieure sont rares. En cas de récidive, le même type de maladie est souvent observé, mais environ 10 % des cas s’aggravent. Si la maladie systémique sous-jacente n’est pas traitée, les récidives surviennent fréquemment au même site de la sclère. Environ 10 % des sclérites nodulaires évoluent vers une sclérite nécrosante au cours de l’évolution.
Un type particulier de sclérite nécrosante avec peu de symptômes inflammatoires est appelé scléromalacie perforante (scleromalacia perforans). Elle survient fréquemment chez les patients atteints de polyarthrite rhumatoïde de longue durée, et un amincissement scléral lent progresse sans rougeur ni douleur. Bien que le nom anglais comporte « perforans » (perforation), la forme du globe oculaire est souvent maintenue par une fine membrane fibreuse.
QQuelle est la différence entre l'épisclérite et la sclérite ?
A
L’épisclérite est une inflammation des plexus vasculaires superficiels, comme le plexus de la capsule de Tenon. La congestion est légère, indolore et n’affecte pas la vision. La sclérite est une inflammation des vaisseaux profonds, accompagnée de douleurs oculaires intenses et d’une congestion rouge foncé. L’instillation d’épinéphrine diluée à 1/1000 fait disparaître la congestion de l’épisclérite, mais pas celle de la sclérite, ce qui permet de les différencier.
Smeller L, Toth-Molnar E, Sohar N. Optical Coherence Tomography: Focus on the Pathology of Macula in Scleritis Patients. J Clin Med. 2023;12(14):4825. Figure 1. PMID: 37510941; PMCID: PMC10381547; DOI: 10.3390/jcm12144825. License: CC BY 4.0.
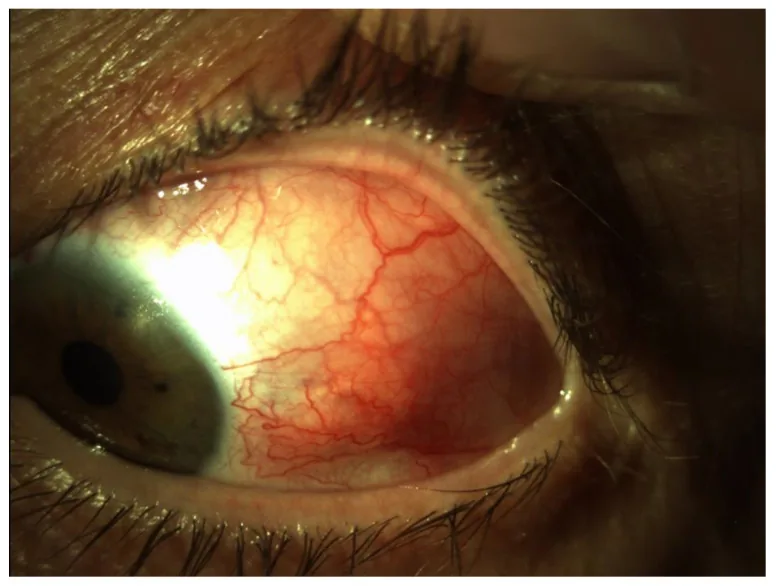
Image du segment antérieur montrant une congestion diffuse avec dilatation marquée et tortuosité des vaisseaux profonds de la sclère du côté temporal de l’œil gauche. Elle correspond aux schémas de congestion et aux signes vasculaires des différents types de sclérite traités dans la section « 2. Principaux symptômes et signes cliniques ».
Douleur oculaire intense : Douleur profonde de type forant (boring), caractéristique. Elle peut être suffisamment sévère pour perturber le sommeil.
Douleur irradiante : La douleur irradie vers l’oreille, le visage, la mâchoire et la tempe. Elle est particulièrement marquée dans la sclérite diffuse.
Aggravation nocturne et douleur aux mouvements oculaires : La douleur s’aggrave la nuit et avec les mouvements oculaires.
Sensibilité à la palpation : Les patients signalent souvent une douleur à la palpation.
Congestion : Le patient ressent une congestion accompagnée d’une douleur intense et pulsatile.
Baisse de l’acuité visuelle : Elle n’est souvent ressentie que dans les cas graves ayant évolué vers une sclérite nécrosante, ou lorsque la rétine ou le nerf optique sont endommagés dans la sclérite postérieure.
Particularité de la scléromalacie perforante : Elle se manifeste par l’apparition soudaine d’un foyer de nécrose sclérale dans un œil sans inflammation, congestion ni douleur, ou par la découverte d’une exposition uvéale due à une perte de substance sclérale.
Signes cliniques (observés par le médecin lors de l’examen)
Dilatation et tortuosité des vaisseaux profonds : L’inflammation des vaisseaux scléraux entraîne une dilatation des plexus vasculaires épiscléraux et intrascléraux. Les vaisseaux scléraux sont immobiles.
Teinte violacée : C’est un changement de couleur caractéristique de la sclérite. Contrairement au rouge vif de la conjonctivite ou de l’épisclérite, la sclérite présente une teinte rouge foncé à violacée. Elle est plus facilement observable à l’œil nu sous lumière naturelle qu’au microscope à lampe à fente. Dans les cas de longue durée, un amincissement scléral localisé ou diffus donne un aspect bleu-noir.
Test à l’épinéphrine : L’instillation d’épinéphrine diluée au 1/1000 ne fait pas disparaître l’hyperhémie des vaisseaux profonds. L’hyperhémie conjonctivale et épisclérale superficielle disparaît, ce qui est utile pour différencier la sclérite de la conjonctivite ou de l’épisclérite.
Absence de signes conjonctivaux palpébraux : Même dans les cas graves, il n’y a pas de signes inflammatoires de la conjonctive palpébrale, ce qui facilite la distinction avec la conjonctivite.
Douleur à la palpation : C’est un élément important pour différencier la sclérite de la conjonctivite ou de l’épisclérite.
Différences selon le type : Chaque type présente des caractéristiques distinctes, généralement identifiables par l’examen à la lampe à fente lors de la première consultation.
Sclérite diffuse
Hyperhémie : Dilatation et tortuosité des vaisseaux scléraux entraînant une hyperhémie intense et diffuse. Elle peut être circonférentielle ou localisée à un quadrant ou plus.
Douleur : Douleur intense irradiant vers le visage ou la tempe, pouvant perturber le sommeil.
Particularités : Absence de nodules, de soulèvements, de nécrose ou d’amincissement. Un œdème conjonctival, un gonflement palpébral, une uvéite antérieure ou une hypertension oculaire peuvent être associés.
Sclérite nodulaire
Nodules : Nodules rouge foncé, uniques ou multiples, apparaissant de préférence près du limbe dans la zone palpébrale.
Palpation : Les nodules sont fixes et douloureux à la palpation.
Antécédents : Souvent associés à un zona ophtalmique antérieur. Environ 10 % évoluent vers une sclérite nécrosante, mais un traitement précoce permet la guérison avec seulement de petites cicatrices.
Sclérite nécrosante
Nécrose sclérale : Au début, zones avasculaires blanc-jaunâtre localisées (foyers de nécrose sclérale). Dilatation et tortuosité marquées des vaisseaux scléraux, avec fonte tissulaire.
Amincissement : Amincissement jusqu’à laisser voir la choroïde, pouvant évoluer vers une perforation oculaire. L’amincissement persiste même après la résolution de l’inflammation.
Pronostic : L’âge d’apparition est élevé, autour de 60 ans, et environ 60 % des cas sont bilatéraux. Sans traitement précoce, la cécité et la difficulté à conserver le globe oculaire sont inévitables.
Sclérite postérieure
Épidémiologie : L’âge moyen d’apparition est d’environ 50 ans, avec une prédominance féminine (environ deux fois plus que les hommes). La bilatéralité est de 30 à 40 %.
Signes du fond d’œil : Œdème papillaire, plis choroïdiens, décollement de rétine exsudatif, masse sous-rétinienne, hypertension oculaire. Des cas d’épanchement uvéal et de glaucome secondaire par fermeture de l’angle ont également été rapportés.
Symptômes d’extension : En cas de myosite des muscles extra-oculaires associée, on observe une diplopie, une douleur à la mobilisation oculaire, une exophtalmie et une ptose palpébrale.
Lorsque l’inflammation s’étend de la sclère à la cornée, elle provoque des infiltrations limbiques et des ulcères. Une uvéite antérieure peut également être associée. La sclérite s’accompagne presque toujours d’une inflammation de l’épisclère, de sorte que les signes d’épisclérite sont également présents.
La sclérite postérieure est généralement diagnostiquée tardivement car les lésions sont situées profondément dans le fond d’œil. Elle peut survenir simultanément avec une sclérite antérieure ou de manière décalée dans le temps. Environ un tiers des sclérites postérieures sont associées à une sclérite antérieure, et environ 70 % des patients développent une sclérite antérieure au cours de l’évolution de la sclérite postérieure1). Les cas avec sclérite antérieure sont plus fortement associés à des maladies systémiques.
Décollement de rétine exsudatif : Décollement séreux de la rétine au pôle postérieur1).
Œdème papillaire : Apparaît lorsque l’inflammation s’étend aux tissus orbitaires ou au nerf optique, nécessitant un traitement urgent pour éviter une perte visuelle permanente.
Signe T à l’échographie en mode B : En raison de l’épaississement scléral et de l’accumulation de liquide dans l’espace de Tenon, la limite entre le nerf optique et la sclère apparaît anguleuse1). C’est le signe échographique le plus spécifique de la sclérite postérieure.
Confusion avec une tumeur choroïdienne : La sclérite postérieure peut être adressée comme une masse choroïdienne et constitue une cause de pseudomélanome1).
QPourquoi la sclérite postérieure est-elle souvent négligée ?
A
Dans la sclérite postérieure, les signes du segment antérieur sont rares ; certains patients consultent uniquement pour une douleur oculaire, des maux de tête ou une baisse de l’acuité visuelle. Les plis choroïdiens et le décollement exsudatif de la rétine, signes du fond d’œil, sont souvent diagnostiqués avec certitude seulement après l’évaluation du signe T à l’échographie en mode B et de l’épaississement choroïdien par OCT1). Il peut également être confondu avec une tumeur choroïdienne, d’où l’importance d’un diagnostic différentiel exhaustif.
Jusqu’à 50 % des sclérites sont associées à une maladie auto-immune systémique. Si la maladie systémique sous-jacente n’est pas traitée, des récidives au même site scléral sont fréquentes. Les causes de la sclérite nécrosante comprennent souvent les maladies rhumatismales, les vascularites et les maladies hématologiques.
Collagénoses et maladies rhumatismales
Polyarthrite rhumatoïde (PR) : C’est la maladie systémique la plus fréquemment associée. Elle peut provoquer une sclérite nécrosante ou une scléromalacie perforante. Elle est typique chez les patients sous traitement à long terme.
Lupus érythémateux disséminé (LED) : Peut être associé à une sclérite antérieure.
Polychondrite chronique atrophiante : Peut être associée à une sclérite antérieure et postérieure, avec des rémissions et des exacerbations.
Vascularites et autres
Granulomatose avec polyangéite (GPA) : Associée à une sclérite nécrosante et une scléromalacie perforante, avec une évolution sévère. En tant que vascularite associée aux ANCA, l’atteinte oculaire peut être le premier symptôme 3).
Artérite de Takayasu : Rarement associée à une sclérite, elle peut être l’occasion de diagnostiquer une vascularite systémique 4).
D’autres associations ont été rapportées : sarcoïdose, maladie de Behçet, maladie de Crohn, rectocolite hémorragique, rhumatisme psoriasique, sclérodermie, dermatomyosite, syndrome SAPHO, maladies thyroïdiennes, syndrome de l’aorte, néphrite interstitielle, maladie de Vogt-Koyanagi-Harada, sclérose en plaques, etc. Dans la sclérite nodulaire, on retrouve souvent des antécédents de zona ophtalmique. Dans la sclérite postérieure, il faut être attentif car elle peut rarement être une manifestation oculaire d’un lymphome systémique ou d’un myélome multiple.
La sclérite infectieuse ne représente que 5 à 10 % de l’ensemble des cas, mais son pronostic est extrêmement défavorable 7). Environ 50 % des patients atteints de sclérite infectieuse perdent la vision fonctionnelle et environ 27 % subissent une énucléation ou une éviscération 7).
Pseudomonas aeruginosa (Pseudomonas aeruginosa) : C’est l’agent pathogène le plus courant en Europe et en Amérique du Nord 7). La nécrose sclérale progresse rapidement, présentant l’aspect d’une scléromalacie purulente.
Genre Nocardia : Survient après un traumatisme ou chez les patients immunodéprimés 2). L’évolution chronique avec des rémissions et des rechutes est caractéristique, et même après la résolution des nodules, les bactéries persistent en profondeur 2).
Genre Moraxella : Agent pathogène rare, mais survient comme infection opportuniste en cas d’immunodépression 7).
Autres : Des cas d’infection par des champignons, la tuberculose, la syphilis, le virus de l’herpès, etc., ont également été rapportés. Dans les régions à forte prévalence de tuberculose, il est recommandé d’exclure la tuberculose par un test tuberculinique avant l’administration systémique de corticostéroïdes.
La plupart des sclérites infectieuses sont causées par des fils de suture exposés ou du matériel de boucle sclérale après une chirurgie oculaire, et surviennent de manière unilatérale.
Sclérite nécrosante induite par la chirurgie (SINS)
Une sclérite nécrosante peut survenir à la suite d’une chirurgie oculaire. Les interventions typiques sont la chirurgie du ptérygion, la chirurgie de la cataracte, la mise en place d’une boucle sclérale, la chirurgie du strabisme et la trabéculectomie. Elle est particulièrement fréquente après une excision du ptérygion avec mitomycine C. Le délai d’apparition varie de quelques jours à plusieurs années après la chirurgie, et les cas survenant plusieurs années après l’opération ne sont pas rares.
Les antimétabolites mitomycine C (MMC) et 5-fluorouracile (5-FU) ont été utilisés pour prévenir la récidive du ptérygion et la cicatrisation des bulles de filtration glaucomateuses. L’utilisation de collyre à la MMC a été abandonnée dans les années 1980 car elle pouvait entraîner une calcification sclérale ou une scléromalacie perforante plusieurs mois à plusieurs années après l’opération. Actuellement, dans la chirurgie du glaucome et du ptérygion, on utilise principalement une application unique peropératoire de MMC à faible concentration (0,02-0,04 %) pendant une courte durée. Cependant, une pâleur sclérale, un rétrécissement vasculaire et des zones avasculaires peuvent apparaître au site chirurgical après l’opération, ce qui peut prédisposer à une scléromalacie future.
QQuelles maladies systémiques sont associées ?
A
La polyarthrite rhumatoïde est la plus fréquente, suivie par la granulomatose avec polyangéite (GPA), le lupus érythémateux disséminé, la périartérite noueuse, la polychondrite atrophiante, l’artérite de Takayasu et la sarcoïdose. Jusqu’à 50 % des patients atteints de sclérite présentent une maladie systémique. Dans la sclérite nécrosante, la fréquence des maladies rhumatismales, des vascularites et des maladies hématologiques est encore plus élevée.
Observation à l’œil nu sous lumière naturelle : Contrairement à la rougeur vive de la conjonctivite ou de l’épisclérite, la sclérite présente une teinte rouge foncé à violet bleuâtre. Dans les cas de longue durée, l’amincissement scléral donne un aspect bleu-noir. Ces changements de couleur sont plus facilement perceptibles à l’œil nu en salle claire qu’au microscope à lampe à fente.
Examen à la lampe à fente : Évalue la dilatation et la tortuosité des vaisseaux scléraux, la présence de nodules, l’aspect rouge foncé des nodules scléraux, l’amincissement, la nécrose et la perforation. L’absence de signes inflammatoires sur la conjonctive palpébrale est un point de différenciation avec la conjonctivite.
Test à l’épinéphrine : L’instillation d’épinéphrine diluée au 1/1000 ne fait pas disparaître l’hyperhémie sclérale profonde. Important pour différencier l’épisclérite et l’hyperhémie conjonctivale.
Palpation : Vérifier la présence de douleur à la pression en touchant la conjonctive avec un coton-tige. Aide à la différenciation avec la conjonctivite et l’épisclérite.
Échographie en mode B : Indispensable pour le diagnostic de la sclérite postérieure. L’épaississement scléral, les nodules scléraux et le signe en T dû à l’accumulation de liquide sous la capsule de Tenon sont caractéristiques 1). Utile également pour différencier les tumeurs choroïdiennes.
Angiographie sclérale à la fluorescéine : Permet de différencier la sclérite nécrosante par la présence de zones de non-perfusion sclérale.
CT et IRM : Utilisés pour évaluer l’épaississement scléral dans la sclérite postérieure, la myosite des muscles extra-oculaires et pour différencier les lésions intracrâniennes.
Infections : Sérologie syphilitique, Quantiféron, test tuberculinique
Autres : Enzyme de conversion de l’angiotensine (ECA), lysozyme, acide urique sérique
Dans la sclérite nécrosante et les cas réfractaires, la recherche d’une vascularite associée aux ANCA est particulièrement importante 3). Dans les régions à forte prévalence de tuberculose, un test tuberculinique doit être effectué avant l’administration systémique en cas de sclérite résistant au traitement stéroïdien local.
Épisclérite : inflammation des vaisseaux superficiels, avec hyperémie légère, absence de douleur, et régression après instillation de phényléphrine.
Conjonctivite : l’hyperémie est la plus marquée au niveau du fornix conjonctival et diminue en approchant du limbe. Elle s’accompagne de sécrétions oculaires et d’anomalies de la conjonctive palpébrale.
Lymphome MALT : tumeur rose saumon survenant fréquemment au niveau du fornix conjonctival. Elle se distingue de la sclérite par le fait que les vaisseaux scléraux ne sont pas visibles en raison de la localisation sous-conjonctivale de la tumeur.
Maladies cornéennes : il est nécessaire de différencier l’infiltrat cornéen périphérique secondaire à une sclérite de l’ulcère de Mooren ou de l’infiltrat cornéen périphérique staphylococcique.
Ténonite : considérée comme un type d’épisclérite, la distinction entre les deux est difficile.
Syndrome de l’apex orbitaire : dans la fistule carotido-caverneuse, on observe une congestion et une dilatation des veines conjonctivales et sclérales, accompagnées d’exophtalmie pulsatile et de diplopie.
Maladie de Vogt-Koyanagi-Harada : difficile à différencier de la sclérite postérieure. Elle se présente sous forme bilatérale avec une uvéite antérieure granulomateuse et un épaississement choroïdien à l’OCT.
Tumeur choroïdienne : les lésions nodulaires de la sclérite postérieure peuvent être référées comme des masses choroïdiennes 1) ; la différenciation se fait par une combinaison d’échographie, d’IRM et d’OCT.
Le traitement de la sclérite repose principalement sur les stéroïdes, avec une combinaison progressive de traitement local, systémique, d’immunosuppresseurs, de biothérapies et de traitement chirurgical en fonction du type et de la sévérité. En cas de maladie systémique associée, une collaboration avec un rhumatologue ou un interniste spécialisé en maladies auto-immunes est indispensable.
AINS oraux (première intention)
Indications : traitement initial de la sclérite diffuse ou nodulaire légère à modérée.
Exemple de prescription : célécoxib (inhibiteur de la COX-2) 100 mg deux fois par jour, ou indométacine 50 mg trois fois par jour. Souvent très efficace contre la douleur et également utile pour contrôler l’inflammation.
Attention : surveiller les saignements gastro-intestinaux, l’insuffisance rénale et les crises d’asthme. En l’absence de contre-indications telles que l’asthme, utiliser en association de manière proactive dès le début.
Traitement local par corticoïdes
Collyre : utiliser du bétaméthasone phosphate disodique à 0,1 % en collyre 4 à 6 fois par jour. Selon les cas, associer une pommade à la bétaméthasone et à la fradiomycine en instillation au coucher.
Injection sous-conjonctivale : triamcinolone acétonide 40 mg/mL, 0,1 mL par injection (jusqu’à une fois par mois), ou dexaméthasone 3,3 mg/mL, 0,3 mL par injection, toutes les 1 à 2 semaines pour quelques injections.
Attention : en cas de sclérite nécrosante, éviter les zones amincies lors de l’injection.
Traitement systémique par corticoïdes
Voie orale : prednisolone 0,5 à 1 mg/kg/jour (en cas d’échec des AINS dans les formes légères, 20 à 30 mg en deux prises avec réduction progressive ; dans les formes graves nodulaires, nécrosantes ou postérieures, 30 à 60 mg/jour avec réduction progressive).
Traitement par bolus : méthylprednisolone 1 000 mg/jour en perfusion intraveineuse pendant 3 jours, suivi d’une réduction progressive. Indiqué dans la sclérite nécrosante et les cas graves.
Attention : la réduction progressive se fait généralement sur 1 à 2 semaines ou plus, et dans les cas graves, elle se poursuit sur 2 à 3 mois.
Immunosuppresseurs et agents biologiques
Ciclosporine : débuter à 5 mg/kg/jour en deux prises orales, ajuster pour maintenir un taux résiduel sanguin autour de 100 à 150 ng/mL. Surveiller la fonction rénale et effectuer des analyses sanguines régulières.
Choix en cas de maladie systémique associée : pour la polyarthrite rhumatoïde, le méthotrexate est souvent choisi ; pour le lupus érythémateux disséminé ou les vascularites systémiques, le cyclophosphamide est souvent préféré. L’azathioprine est moins efficace dans la sclérite.
Agents biologiques : des rapports font état de l’utilisation de l’infliximab (anticorps anti-TNF-α, Remicade®) et du rituximab (anticorps anti-CD20, Rituxan®) dans les cas réfractaires3).
Le traitement commence par des AINS par voie orale et des gouttes oculaires de bétaméthasone à 0,1 %. Si la réponse est insuffisante, on ajoute des collyres immunosuppresseurs. Si cela reste insuffisant, on effectue une injection sous-conjonctivale de 0,3 mL de dexaméthasone ou de 0,1 à 0,2 mL d’acétonide de triamcinolone, en évitant les zones sclérales amincies. En cas de mauvaise réponse au traitement local, on poursuit une corticothérapie orale dégressive de prednisolone à 20-30 mg/jour pendant 1 à 2 semaines.
En raison du risque d’amincissement et de perforation sclérale à court terme, on commence d’emblée par de la prednisolone orale à 0,5-1 mg/kg/jour. Pour les cas ne répondant pas aux corticostéroïdes oraux ou présentant des rechutes fréquentes, on choisit, en collaboration avec un rhumatologue, l’immunosuppresseur le plus adapté à la maladie systémique associée. En cas d’utilisation concomitante de cyclosporine à 5 mg/kg/jour en deux prises orales, on ajuste la concentration résiduelle sanguine entre 100 et 150 ng/mL. Pour la sclérite associée à la maladie de Behçet neurologique, la cyclosporine est contre-indiquée car elle aggrave les symptômes neurologiques. Dans les cas graves, on réalise une thérapie par bolus de méthylprednisolone à 1 000 mg/jour en perfusion intraveineuse pendant 3 jours, après avoir exclu toute cause infectieuse.
Pour les sclérites résistantes au traitement immunosuppresseur, on envisage l’introduction d’agents biologiques. Les inhibiteurs du TNF-α ont montré leur efficacité dans la sclérouvéite, mais tous ne sont pas efficaces contre la sclérite ; l’étanercept a été associé à des cas de réaction paradoxale induisant ou aggravant une inflammation oculaire, y compris une sclérite. L’utilisation d’immunosuppresseurs et de produits biologiques nécessite des examens systémiques avant et après l’introduction, ainsi qu’une collaboration avec un interniste.
Le traitement principal repose sur l’administration systémique de corticostéroïdes. On commence par de la prednisolone à 30-50 mg/jour en doses fractionnées 2 à 3 fois par jour pour contrôler la douleur, puis on réduit progressivement. En cas d’inflammation du segment antérieur, on associe des collyres corticostéroïdes. Si l’inflammation ne se calme pas avec la prise orale, on effectue un traitement par bolus de stéroïdes après avoir exclu toute cause infectieuse. En cas de résistance au bolus ou de rechute lors de la diminution, on envisage l’administration active d’immunosuppresseurs. Des exemples de prescriptions incluent l’azathioprine à 1-2 mg/kg, le méthotrexate (Rheumatrex®) à 6 mg/semaine. Dans la sclérite nécrosante, un traitement par bolus de cyclophosphamide peut être nécessaire, et la collaboration avec un interniste est particulièrement importante.
L’injection sous-ténonienne d’acétonide de triamcinolone (STTA) est également utilisée pour la sclérite postérieure, mais elle comporte un risque rare de troubles circulatoires du nerf optique et de la choroïde rétinienne6). Chez les personnes âgées à fragilité vasculaire ou les patients atteints de glaucome, une évaluation prudente de l’indication est nécessaire6).
Dans la sclérite infectieuse, le traitement de base est l’antibiothérapie basée sur l’identification de l’agent pathogène et sa sensibilité2)7).
Traitement sélectif après identification de l’agent pathogène : En cas d’infection à Nocardia, associer des gouttes oculaires d’amikacine renforcées et du sulfaméthoxazole-triméthoprime par voie orale pendant une longue période. Un débridement chirurgical répété peut être nécessaire2). En cas d’infection à Pseudomonas aeruginosa, la nécrose sclérale (scléromalacie purulente) progresse rapidement, nécessitant un traitement intensif à base d’aminosides ou de fluoroquinolones et une intervention chirurgicale rapide.
Retrait des sutures exposées et du matériel de boucle : Les sutures exposées qui sont la source de l’infection doivent être retirées immédiatement. Si le matériel de boucle sclérale ne répond pas au traitement médicamenteux, il est souhaitable de le retirer dans un délai d’une à deux semaines pour prévenir une endophtalmie.
Décision d’utiliser des corticostéroïdes en association : En raison du risque d’aggravation de l’infection par les corticostéroïdes, ils ne doivent être utilisés qu’après avoir exclu de manière suffisante une cause infectieuse. Si le patient répond aux antibiotiques mais que l’inflammation persiste, les corticostéroïdes peuvent être utilisés en surveillant la numération leucocytaire et la CRP.
Sclérite fongique : Le traitement suit celui de la kératite fongique.
La sclérite nécrosante ou la scléromalacie perforante, ainsi que la sclérite infectieuse ne répondant pas au traitement médical, sont des indications chirurgicales. Lorsque la nécrose ou la zone de ramollissement de la sclère dépasse une certaine étendue, il devient difficile de restaurer la forme du globe oculaire et de maintenir la fonction visuelle. Une intervention chirurgicale précoce est donc souhaitable tant que la zone nécrosée est petite.
Les points clés de la chirurgie sont les trois suivants :
Résection complète du foyer de nécrose sclérale, y compris les zones saines environnantes
Réparation et comblement de la lésion par greffe de sclère conservée : La sclère conservée est un matériau de comblement approprié pour sa résistance et le maintien de la morphologie de la paroi oculaire. La cornée conservée se lyse souvent.
Recouvrement complet du greffon scléral par la conjonctive
En cas de nécrose conjonctivale étendue ou d’ulcère cornéen périphérique, une autogreffe conjonctivale de l’autre œil ou une greffe épithéliale cornéenne peut être associée. En postopératoire, on utilise de la ciclosporine par voie orale et des gouttes oculaires de Sandimmun® à 1% (préparation hospitalière) pour favoriser la prise du greffon et prévenir la récidive de la sclérite. Même dans la scléromalacie survenue après l’utilisation de MMC ou de 5-FU, en l’absence de preuves solides de l’efficacité des collyres immunosuppresseurs ou des agents biologiques, une greffe précoce de sclère conservée doit être réalisée tant que la zone de ramollissement est petite.
Prise en charge systémique et surveillance des effets secondaires
Lors d’un traitement prolongé par corticostéroïdes et immunosuppresseurs, il est nécessaire de surveiller régulièrement la pression intraoculaire, les fonctions hépatique et rénale, la glycémie et la concentration sanguine de ciclosporine. En cas d’hyperglycémie, un traitement par corticoïdes sous insuline peut être nécessaire avec une prise en charge systémique en collaboration avec un spécialiste en médecine interne. Des analgésiques anti-inflammatoires sont administrés pour la douleur oculaire. De plus, comme la sclérite peut survenir à la suite d’une infection ou d’une allergie infectieuse, un traitement initial par collyres et antibiotiques oraux est également associé.
QComment traite-t-on la sclérite infectieuse ?
A
La sclérite infectieuse a un pronostic sombre ; l’identification de l’agent pathogène et l’administration précoce d’antibiotiques en fonction de la sensibilité sont essentielles. En cas d’infection à Pseudomonas aeruginosa, qui progresse rapidement, un traitement antibiotique puissant et un débridement chirurgical rapide sont nécessaires 7). Si des fils de suture exposés ou du matériel de boucle sont en cause, ils doivent être retirés rapidement. Les stéroïdes risquent d’aggraver l’infection, ils doivent donc être utilisés avec prudence en cas de suspicion d’infection.
La sclère est un tissu fibreux peu vascularisé, et la sclérite profonde est rare. Cependant, comme la sclère est innervée, une fois l’inflammation déclenchée, elle provoque une douleur oculaire intense. L’épaisseur de la sclère au niveau de l’insertion des muscles droits est d’environ 0,3 mm, ce qui en fait un site fréquent de nécrose et de perforation. La sclère ne possède pas de barrière, de sorte que les médicaments injectés sous la conjonctive ou sous la capsule de Tenon peuvent atteindre l’intérieur de l’œil par diffusion, ce qui explique pourquoi les injections sous-conjonctivales de stéroïdes sont une option thérapeutique.
La pathologie de la sclérite associée aux maladies auto-immunes se caractérise par une nécrose granulomateuse zonale. Au centre du granulome se trouve une substance fibrinoïde, entourée de cellules épithélioïdes et de cellules géantes multinucléées.
Dans la sclérite, on observe une augmentation de l’infiltration de cellules inflammatoires, notamment des lymphocytes T et des macrophages. Les lymphocytes T et les macrophages infiltrent le tissu scléral profond, et des amas de lymphocytes B se forment autour des vaisseaux. L’expression accrue de HLA-DR et du récepteur de l’IL-2 sur les lymphocytes T suggère l’implication d’une réponse immunitaire à médiation cellulaire.
Les plasmocytes sont impliqués dans la production de métalloprotéinases matricielles (MMP) et de TNF-α. Dans la sclérite nécrosante, on observe une vascularite avec nécrose fibrinoïde et infiltration de neutrophiles dans la paroi vasculaire. Le mécanisme de la sclérite endogène implique probablement une réponse immunitaire à médiation cellulaire.
Sclérite non nécrosante (diffuse ou nodulaire) : la vascularite n’est pas marquée, l’inflammation est principalement non granulomateuse. Dans la forme nodulaire, on observe une nécrose fibrinoïde centrale entourée de cellules épithélioïdes.
Sclérite nécrosante : on observe de petits foyers de nécrose et une inflammation non granulomateuse principalement composée de lymphocytes, de plasmocytes et de macrophages. La vascularite avec nécrose fibrinoïde et infiltration de neutrophiles est caractéristique.
Sclérite infectieuse : en plus de l’inflammation nécrosante, des micro-abcès se forment. Dans l’infection à Nocardia, même après la disparition des nodules, les bactéries persistent en profondeur et les récidives sont fréquentes 2).
Scléromalacie perforante : survient chez les patients atteints de polyarthrite rhumatoïde de longue durée ou de maladies apparentées. Elle se présente sous forme de plaques nécrotiques sclérales sans congestion près du limbe, avec un amincissement scléral lent progressant jusqu’à l’exposition de l’uvée.
Une série de 8 cas de sclérite postérieure survenue après vaccination ou infection COVID-19, initialement confondue avec un mélanome choroïdien, a été rapportée 5). L’intervalle moyen entre la dernière dose de vaccin et l’apparition était de 132 jours, et entre l’infection COVID-19 et l’apparition de 14 jours 5). La plupart des cas se sont résolus spontanément en 2 mois avec un impact visuel minime 5).
Rituximab pour la sclérite réfractaire associée aux ANCA
Des cas de sclérite nécrosante associée aux ANCA résistante à l’immunosuppression conventionnelle (stéroïdes + cyclophosphamide) ont montré une efficacité du rituximab (anticorps anti-CD20) pour l’induction et le maintien de la rémission 3). Des études de suivi à long terme s’accumulent également sur l’efficacité du rituximab dans les lésions oculaires de la granulomatose avec polyangéite associée aux ANCA 3).
Des cas de sclérite comme manifestation oculaire de l’artérite de Takayasu ont été rapportés, nécessitant une attention en tant qu’indice diagnostique du syndrome d’aortite systémique 4). Chez les jeunes femmes présentant une sclérite nécrosante, il est important d’exclure l’artérite de Takayasu.
QY a-t-il un lien avec le COVID-19 ?
A
Des séries de cas de sclérite postérieure après vaccination ou infection COVID-19 ont été rapportées 5). Cependant, la relation causale n’est pas prouvée et la plupart des cas évoluent vers une résolution spontanée. La sclérite postérieure liée au COVID-19 peut être confondue avec une tumeur choroïdienne, d’où l’importance de la reconnaître comme diagnostic différentiel 5).
Babu N, Kumar K, Upadhayay A, Kohli P. Nodular posterior scleritis - The great masquerader. Taiwan journal of ophthalmology. 2021;11(4):408-412. doi:10.4103/tjo.tjo_20_21. PMID:35070674; PMCID:PMC8757530.
Chauhan K, Murthy SI, Mitra S. Demystifying nocardial scleritis. BMJ case reports. 2023;16(11). doi:10.1136/bcr-2023-255730. PMID:38011958; PMCID:PMC10685915.
Tahavvori M, Fekri S, Hassanpour K, Sadoughi MM, Javadi M. Isolated ANCA-associated scleritis successfully treated with systemic rituximab; a case report and review of literature. BMC ophthalmology. 2025;25(1):176. doi:10.1186/s12886-025-04027-6. PMID:40197146; PMCID:PMC11974155.
Chittipolu S, Kennard JL, Tumma RS, Doyle AR. Scleritis in Takayasu Arteritis. Cureus. 2023;15(4):e37724. doi:10.7759/cureus.37724. PMID:37206528; PMCID:PMC10191460.
Negretti GS, Zeiger JS, Cherkas E, Shields CL. Posterior scleritis following COVID-19 vaccination or infection simulating uveal melanoma in 8 consecutive patients. Eye (London, England). 2024;38(1):185-191. doi:10.1038/s41433-023-02656-z. PMID:37422535; PMCID:PMC10764359.
Akada M, Muraoka Y, Morooka S, Ishihara K, Hata M, Tsujikawa A. SEVERE CIRCULATORY DISTURBANCE IN OPTIC DISK, RETINA, AND CHOROID AFTER SUB-TENON TRIAMCINOLONE ACETONIDE INJECTION FOR POSTERIOR SCLERITIS. Retinal cases & brief reports. 2025;19(6):789-792. doi:10.1097/ICB.0000000000001642. PMID:39058980; PMCID:PMC12570599.
Dallinga M, Murtagh P, Powell S, Murphy CC. Moraxella nonliquefaciens-associated infectious scleritis. BMJ case reports. 2023;16(5). doi:10.1136/bcr-2022-254113. PMID:37221000; PMCID:PMC10230883.