SEP-RR
Forme récurrente-rémittente (SEP-RR) : le sous-type le plus fréquent. Les poussées durent plus de 24 heures et sont suivies d’une rémission complète ou partielle.

La sclérose en plaques (SEP) est une maladie inflammatoire démyélinisante de la substance blanche du système nerveux central (SNC), caractérisée par des symptômes neurologiques variés évoluant par poussées et rémissions. Elle se distingue par des lésions sclérotiques cicatricielles dues à la gliose, et touche généralement uniquement le SNC, épargnant le système nerveux périphérique.
La prévalence estimée aux États-Unis est de 1 à 1,5 pour 1 000 personnes1). Dans le monde, 2,1 millions de personnes sont atteintes, avec une distribution plus fréquente dans les régions de haute latitude de l’hémisphère nord et sud. L’âge moyen d’apparition se situe entre 15 et 45 ans, et l’âge moyen au diagnostic est de 30 ans. La tranche d’âge la plus touchée est de 15 à 50 ans, avec une prédominance féminine (pic vers la fin de la vingtaine), et un sex-ratio de 1:2,9.
La SEP comporte quatre sous-types principaux. La SEP-RR (poussées-rémissions) débute généralement entre 25 et 29 ans, tandis que la SEP-SP survient souvent entre 40 et 49 ans1).
SEP-RR
Forme récurrente-rémittente (SEP-RR) : le sous-type le plus fréquent. Les poussées durent plus de 24 heures et sont suivies d’une rémission complète ou partielle.
SEP-SP
Forme secondairement progressive (SEP-SP) : évolution de la SEP-RR. Accumulation progressive de handicaps même pendant les phases de rémission.
SEP-PP
Forme primaire progressive (SEP-PP) : accumulation progressive de handicaps dès le début, sans poussées, avec une progression lente.
CIS
Syndrome cliniquement isolé (CIS) : premier épisode clinique pouvant évoluer vers la SEP. Permet un traitement précoce.
La SEP se divise en quatre sous-types : SEP-RR (récurrente-rémittente), SEP-SP (secondairement progressive), SEP-PP (primaire progressive) et CIS (syndrome cliniquement isolé). Le plus fréquent est la SEP-RR, caractérisée par des poussées et des rémissions. La SEP-SP succède à la SEP-RR, tandis que la SEP-PP progresse dès le début.
Chez 75% des patients, le premier symptôme est une plainte unique, 45% présentent des symptômes moteurs/sensoriels et 20% des symptômes visuels.
Symptômes oculaires
Symptômes neurologiques généraux
L’exacerbation est aiguë à subaiguë et dure de quelques jours à quelques mois. Dans 85 % des cas, les symptômes s’améliorent ou disparaissent, mais des séquelles persistent dans 10 à 15 % des cas.
Elle se manifeste souvent par une baisse de l’acuité visuelle unilatérale douloureuse. Une douleur orbitaire est observée dans 92 % des cas, aggravée par les mouvements oculaires. On observe également le phénomène d’Uhthoff, où une augmentation de la température corporelle (bain, exercice) aggrave temporairement les symptômes.
La cause exacte de la SEP est inconnue, mais on pense qu’un mécanisme auto-immun est impliqué dans son apparition.
Les facteurs génétiques sont impliqués, mais le taux de concordance chez les jumeaux monozygotes n’est que de 25 à 30 %. Bien que le polymorphisme HLA et plus de 100 loci de risque aient été identifiés, on pense que non seulement la prédisposition génétique mais aussi les facteurs environnementaux jouent un rôle important dans l’apparition de la maladie.
Les critères de McDonald 2017 (révision 2024) sont utilisés. La démonstration de la dissémination temporelle et spatiale (DIT/DIS) des lésions démyélinisantes du système nerveux central est fondamentale. Dans la révision de 2024, le nerf optique a été ajouté comme cinquième site topographique. Au Japon, il existe également les critères diagnostiques de la sclérose en plaques du ministère de la Santé, du Travail et des Affaires sociales de 2015.
Les cinq sites topographiques de la dissémination spatiale (DIS) sont les suivants :
Preuve de dissémination temporelle (DIT) : ≥2 attaques, ou présence simultanée de lésions rehaussées et non rehaussées à l’IRM, nouvelles lésions T2, ou bandes oligoclonales dans le LCR 1).
Pour le diagnostic de PPMS, en plus d’une progression du handicap ≥1 an, au moins 2 des éléments suivants sont nécessaires : lésions T2 cérébrales, lésions T2 médullaires (≥2), ou bandes oligoclonales dans le LCR 1).
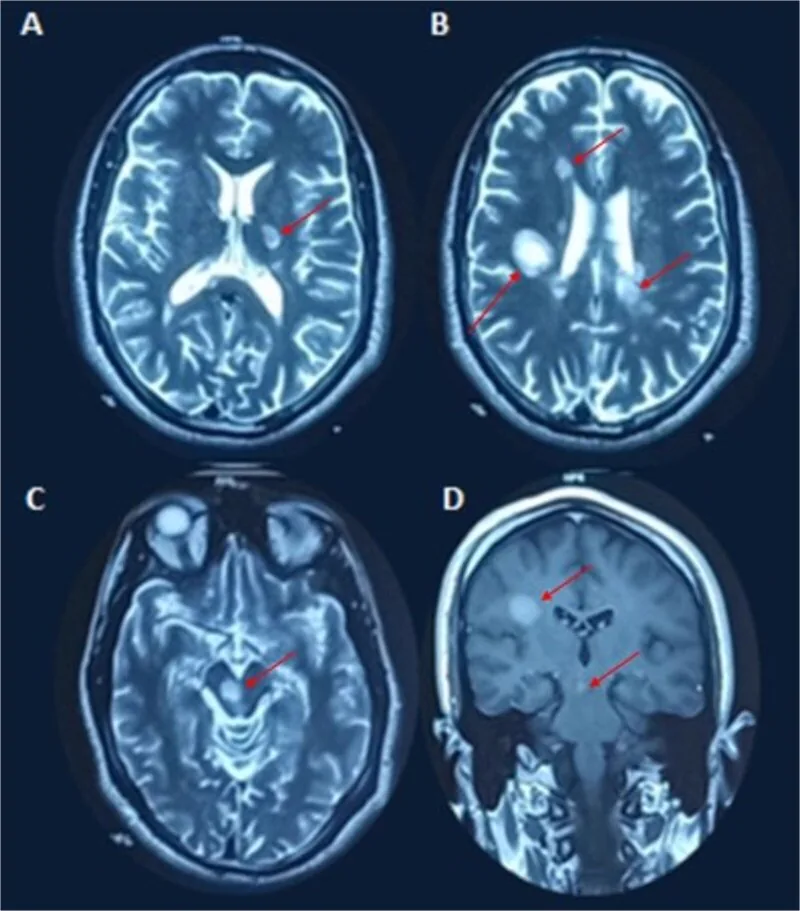
Les plaques de démyélinisation sont détectées comme des lésions hyperintenses en T2 ou des lésions rehaussées par le gadolinium.
Utile lorsque l’IRM est non concluante ou pour prédire la progression de la maladie 1). Peut détecter une démyélinisation précoce et asymptomatique avant qu’elle ne soit visible à l’IRM. Un allongement de la latence et une diminution de l’amplitude sont observés dans 65% des cas.
Il est important de différencier les maladies suivantes ; en cas de présentation atypique, des examens complémentaires sont réalisés.
| Catégorie de maladie | Principaux diagnostics différentiels |
|---|---|
| Maladies démyélinisantes | NMO (maladie de Devic), ADEM, MOGAD |
| Infectieux | Sarcoïdose, tuberculose, syphilis, maladie de Lyme |
| Auto-immun | Lupus érythémateux disséminé (LED), syndrome de Gougerot-Sjögren, maladie de Behçet |
| Maladies du nerf optique | NAION, LHON, neuropathie optique toxique/métabolique |
Examens complémentaires en cas de présentation atypique : anticorps anti-AQP4 (pour exclure une NMO), anticorps anti-MOG (pour exclure une MOGAD), dosage du NfL sérique, sérologie syphilitique (VDRL/RPR/FTA-ABS), ANA (pour le LED), ACE et lysozyme (pour la sarcoïdose).
Le traitement standard au Japon est la corticothérapie par bolus de méthylprednisolone à raison de 1 000 mg/jour en perfusion intraveineuse pendant 3 jours consécutifs. Aucune corticothérapie orale de relais (prednisolone) n’est administrée après la perfusion de 3 jours. La corticothérapie orale augmenterait le taux de récidive et ne doit pas être utilisée.
Même sans traitement, environ 80 % des patients constatent une amélioration de l’acuité visuelle dans les 3 semaines suivant l’apparition des symptômes, mais le traitement par bolus raccourcit la période d’amélioration. On peut s’attendre à une récupération visuelle chez plus de 90 % des cas de névrite optique.
Si le traitement par bolus de corticostéroïdes est inefficace, une épuration extra-rénale (échange plasmatique) est réalisée. À l’étranger, on utilise de la méthylprednisolone à raison de 500 à 1 000 mg/jour pendant 3 à 5 jours. L’essai clinique ONTT (Optic Neuritis Treatment Trial) a montré que la méthylprednisolone intraveineuse à haute dose améliorait le temps de récupération de la fonction visuelle, de la sensibilité au contraste et de la vision des couleurs, mais n’a pas démontré d’amélioration du pronostic visuel final.
Après l’amélioration de la baisse d’acuité visuelle et des troubles du champ visuel, un traitement de fond (DMT) doit être envisagé en collaboration avec un neurologue pour prévenir les récidives.
Les principaux DMT et leur efficacité sont présentés ci-dessous.
| Médicament | Mécanisme d’action | Voie d’administration | Réduction du risque relatif |
|---|---|---|---|
| Interféron bêta | Modulation de l’activité des lymphocytes T/B et de la sécrétion de cytokines | Auto-injection | RR de progression du handicap 0,71 |
| Acétate de glatiramère | Régulation des lymphocytes T régulateurs | Auto-injection | RR de rechute 0,82 |
| Natalizumab | Inhibition de l’infiltration des cellules inflammatoires dans le SNC | Perfusion | RR de rechute 0,56 |
| Fingolimod | Modulateur des récepteurs S1P | Oral | Nouvelles lésions T2 RR 0,65 |
| Tériflunomide | Inhibition de la synthèse des pyrimidines | Oral | Progression du handicap RR 0,76 |
| Diméthylfumarate | Réduction du stress oxydatif et de l’inflammation | Oral | Rechute RR 0,64 |
| Alemtuzumab | Anticorps monoclonal anti-CD52 | Perfusion | Progression du handicap RR 0,44 |
Les anticorps monoclonaux anti-CD20 (ocrelizumab, rituximab, ofatumumab) sont devenus un traitement standard de la SEP récurrente3).
Même en l’absence de lésions cérébrales, 25 % des patients atteints de névrite optique développent une SEP après 15 ans, et 78 % en présence de lésions cérébrales.
Même sans lésion à l’IRM cérébrale, 25 % des patients développent une SEP après 15 ans ; avec lésions, 78 % évoluent vers la SEP. Les patients ayant présenté une névrite optique doivent être suivis en neurologie pour envisager un DMT préventif.
La SEP est considérée comme une maladie auto-immune. Les lymphocytes T reconnaissent la myéline comme étrangère et activent les macrophages, cytokines et anticorps pour détruire la myéline et les axones. La perte de myéline perturbe la conduction de l’influx électrique et altère la transmission des signaux nerveux.
Plaque active
Macrophages spumeux : accumulation de macrophages ayant phagocyté la gaine de myéline.
Infiltration périvasculaire (perivascular cuffing) : aspect caractéristique de lymphocytes entourant les vaisseaux.
Lésions de démyélinisation focales œdémateuses : observées lors des poussées aiguës.
Plaque chronique
Perte de myéline : visible par coloration au Luxol fast blue. Les axones sont préservés mais la remyélinisation est incomplète.
Lésions NAWM : gliose diffuse, activation microgliale et rupture de la BHE dans la substance blanche d’apparence normale. Elles sont plus fortement corrélées aux troubles cliniques que les lésions focales de la substance blanche.
Les oligodendrocytes assurent la remyélinisation du SNC1). Elle dépend des cellules progénitrices d’oligodendrocytes (OPC) adultes, mais les oligodendrocytes matures existants ne peuvent pas contribuer à la remyélinisation1).
Les principales causes d’échec de la remyélinisation sont les suivantes1).
De plus, des lésions de la substance grise corticale et sous-corticale sont également observées, et on sait que la formation de structures lymphoïdes folliculaires à cellules B dans les méninges conduit à une évolution clinique plus sévère 1).
Il s’agit d’une nouvelle approche qui bloque la costimulation entre les cellules T et les cellules présentatrices d’antigènes (y compris les cellules B) en inhibant le CD40L.
Dans l’essai de phase 2 de Vermersch et al. (N Engl J Med 2024), le frexalimab a montré une efficacité claire par rapport au placebo sur les critères IRM (nouvelles lésions rehaussées par le gadolinium à 8-12 semaines), et une diminution du NfL sérique, biomarqueur des lésions du tissu nerveux, a également été confirmée 3). Pour la SEP progressive, on attend également un effet d’inactivation de la microglie et des macrophages, et un blocage du signal CD40L sur la microglie en bordure de plaque pourrait théoriquement permettre une neuroprotection 3).
L’établissement d’un avantage clinique par rapport aux DMT à haute efficacité actuelles (anti-CD20) est considéré comme un défi futur 3).
Il a été démontré que la ferroptose, une mort cellulaire dépendante du fer, est impliquée dans la mort neuronale dans la SEP.
Tang et al. (2025) commentent l’étude de Woo et al. (Cell, 2024) et rapportent une cascade : excitotoxicité du glutamate → surcharge calcique → stress du réticulum endoplasmique → dissociation de STING1 de STIM1 → activation de la voie non canonique → autophagie → dégradation autophagique de GPX4 (enzyme neutralisant la peroxydation lipidique) → ferroptose 4). Une augmentation de l’expression de STING1 dans les neurones a été confirmée à la fois dans des échantillons humains de SEP et dans des modèles murins. Les inhibiteurs de STING1 (C176, H151) ont réduit la dégradation autophagique de GPX4 dans des modèles animaux et ont montré un effet neuroprotecteur 4).
Au stade de la recherche, la neuroprotection via l’inactivation de la microglie et des macrophages par le frexalimab, un inhibiteur de CD40L 3), et l’inhibition de la ferroptose (mort cellulaire dépendante du fer) par l’inhibition de STING1 4) sont considérées comme prometteuses. Les deux sont actuellement en phase d’essai ou de recherche et ne constituent pas un traitement standard.