RRMS
عودکننده-بهبودیابنده (Relapsing-Remitting MS): شایعترین زیرگروه. عودها بیش از ۲۴ ساعت طول میکشند و بین حملات بهبودی کامل یا نسبی دیده میشود.

مولتیپل اسکلروزیس (MS) یک بیماری است که در آن ضایعات دمیلینه کننده التهابی در ماده سفید سیستم عصبی مرکزی (CNS) ایجاد میشود و علائم عصبی متنوعی با الگوی عود و بهبودی بروز میکند. مشخصه آن ضایعات اسکلروتیک ناشی از گلیوز است و معمولاً فقط سیستم عصبی مرکزی درگیر میشود و سیستم عصبی محیطی آسیب نمیبیند.
شیوع تخمینی در ایالات متحده 1 تا 1.5 در 1000 نفر است1). در سراسر جهان 2.1 میلیون نفر مبتلا هستند و در مناطق با عرض جغرافیایی بالا در نیمکره شمالی و جنوبی بیشتر دیده میشود. میانگین سن شروع 15 تا 45 سال و میانگین سن در زمان تشخیص 30 سال است. سن شایع 15 تا 50 سال است و در زنان (با اوج در اواخر دهه 20) شایعتر است و نسبت زن به مرد 1:2.9 است.
MS چهار زیرگروه اصلی دارد: RRMS (عودکننده-بهبودیابنده) معمولاً در سن ۲۵-۲۹ سالگی شروع میشود و SPMS در سن ۴۰-۴۹ سالگی شایعتر است1).
RRMS
عودکننده-بهبودیابنده (Relapsing-Remitting MS): شایعترین زیرگروه. عودها بیش از ۲۴ ساعت طول میکشند و بین حملات بهبودی کامل یا نسبی دیده میشود.
SPMS
پیشرونده ثانویه (Secondary Progressive MS): از RRMS منتقل میشود. در دوره بهبودی نیز ناتوانی به تدریج افزایش مییابد.
PPMS
پیشرونده اولیه (Primary Progressive MS): از ابتدا ناتوانی به تدریج افزایش مییابد. بدون عود به آرامی پیشرفت میکند.
CIS
سندرم ایزوله بالینی (Clinically Isolated Syndrome): اولین رویداد بالینی که میتواند به MS تبدیل شود. امکان شروع زودهنگام درمان را فراهم میکند.
در 75% از بیماران، اولین علامت یک شکایت منفرد است که 45% آن علائم حرکتی/حسی و 20% علائم بینایی است.
علائم چشمی
علائم عصبی عمومی
تشدید به صورت حاد تا تحت حاد شروع شده و چند روز تا چند ماه ادامه مییابد. در ۸۵٪ موارد علائم بهبود یافته یا از بین میروند، اما در ۱۰-۱۵٪ عوارض باقی میماند.
اغلب به صورت کاهش بینایی یک طرفه همراه با درد شروع میشود. درد اربیت در ۹۲٪ موارد وجود دارد و با حرکت چشم تشدید میشود. همچنین پدیده اوهتوف (Uhthoff) که در آن علائم با افزایش دمای بدن (حمام، ورزش) به طور موقت بدتر میشود، دیده میشود.
علت دقیق MS ناشناخته است، اما تصور میشود مکانیسمهای خودایمنی در شروع آن نقش داشته باشند.
عوامل ژنتیکی نقش دارند، اما میزان توافق در دوقلوهای همسان تنها 25 تا 30 درصد است. پلیمورفیسم HLA و بیش از 100 مکان ژنی خطرساز شناسایی شده است، اما تصور میشود که علاوه بر استعداد ژنتیکی، عوامل محیطی نیز نقش مهمی در بروز بیماری دارند.
معیارهای مکدونالد 2017 (نسخه اصلاحشده 2024) استفاده میشود. اثبات پراکندگی زمانی و مکانی (DIT/DIS) ضایعات دمیلینه کننده سیستم عصبی مرکزی اساسی است. در اصلاحیه 2024، عصب بینایی به عنوان پنجمین ناحیه آناتومیک اضافه شد. در ژاپن نیز معیارهای تشخیصی مولتیپل اسکلروزیس وزارت بهداشت، کار و رفاه 2015 وجود دارد.
پنج ناحیه آناتومیک برای پراکندگی مکانی (DIS) به شرح زیر است:
اثبات انتشار زمانی (DIT): دو یا بیش از دو حمله، یا وجود همزمان ضایعات با و بدون افزایش کنتراست در MRI، ضایعات T2 جدید، یا نوارهای الیگوکلونال در CSF میتواند جایگزین شود 1).
برای تشخیص PPMS، علاوه بر پیشرفت ناتوانی به مدت حداقل یک سال، وجود حداقل دو مورد از موارد زیر لازم است: ضایعات T2 مغزی، ضایعات T2 نخاعی (دو یا بیشتر)، یا نوارهای الیگوکلونال در CSF 1).
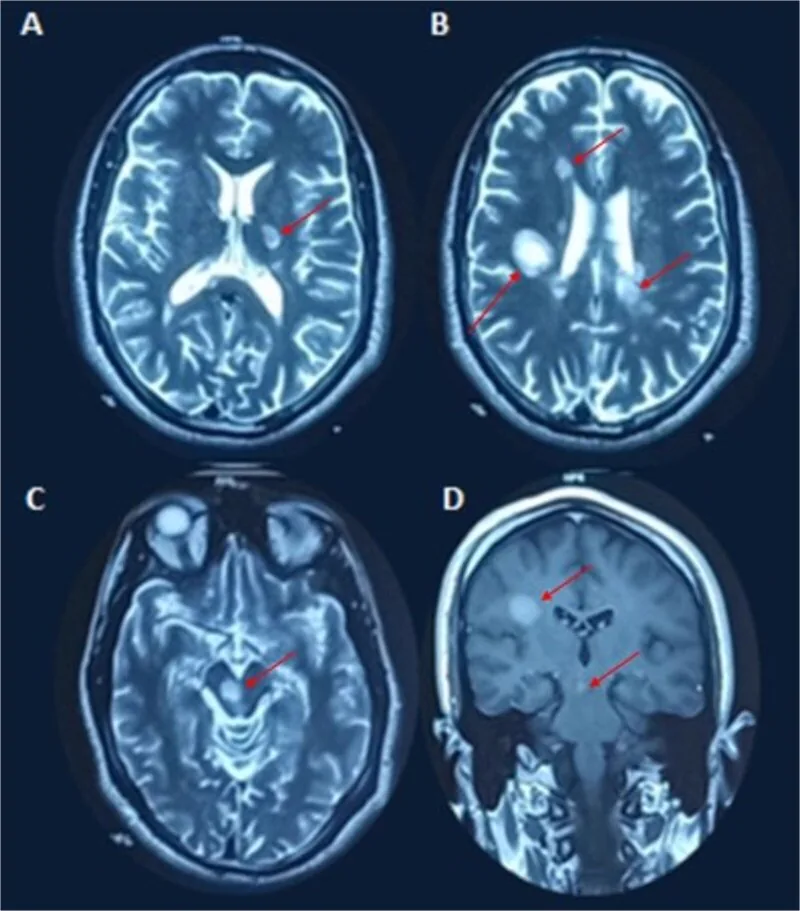
پلاکهای دمیلیناسیون به صورت ضایعات با سیگنال بالا در T2 یا ضایعات با افزایش کنتراست گادولینیوم تشخیص داده میشوند.
در مواردی که MRI قطعی نیست یا برای پیشبینی پیشرفت بیماری مفید است 1). میتواند دمیلیناسیون زودرس و بدون علامت را قبل از قابل مشاهده شدن در MRI تشخیص دهد. در 65% موارد افزایش تأخیر و کاهش دامنه مشاهده میشود.
تشخیص افتراقی با بیماریهای زیر مهم است و در موارد غیرمعمول آزمایشات تکمیلی انجام میشود.
| دسته بیماری | بیماریهای افتراقی اصلی |
|---|---|
| بیماریهای دمیلینهکننده | NMO (بیماری دویک)، ADEM، MOGAD |
| عفونی | سارکوئیدوز، سل، سیفلیس، بیماری لایم |
| خودایمنی | لوپوس اریتماتوز سیستمیک (SLE)، سندرم شوگرن، بیماری بهجت |
| بیماریهای عصب بینایی | NAION، LHON، نوروپاتی بینایی سمی-متابولیک |
آزمایشهای تکمیلی در موارد غیرمعمول: آنتیبادی ضد AQP4 (برای رد NMO)، آنتیبادی ضد MOG (برای رد MOGAD)، آزمایش NfL سرم، آزمایش سرولوژی سیفلیس (VDRL/RPR/FTA-ABS)، ANA (برای SLE)، ACE و لیزوزیم (برای سارکوئیدوز).
درمان استاندارد در ژاپن، پالس استروئیدی با متیلپردنیزولون ۱۰۰۰ میلیگرم در روز به صورت انفوزیون وریدی به مدت ۳ روز متوالی است. پس از ۳ روز انفوزیون، پردنیزولون خوراکی (درمان نگهدارنده) انجام نمیشود. درمان خوراکی استروئیدی خطر عود را افزایش میدهد و نباید انجام شود.
حتی بدون درمان، حدود ۸۰٪ موارد در عرض ۳ هفته از شروع علائم بهبود بینایی را نشان میدهند، اما پالس درمانی دوره بهبود را کوتاهتر میکند. در بیش از ۹۰٪ موارد نوریت بینایی، بهبود بینایی قابل انتظار است.
در صورت عدم پاسخ به پالس استروئیدی، از پلاسمافرز (تعویض پلاسما) استفاده میشود. در خارج از ژاپن، متیلپردنیزولون ۵۰۰-۱۰۰۰ میلیگرم در روز به مدت ۳-۵ روز استفاده میشود. در مطالعه درمان نوریت بینایی (ONTT)، انفوزیون وریدی با دوز بالای متیلپردنیزولون زمان بهبود عملکرد بینایی، حساسیت کنتراست و دید رنگی را بهبود بخشید، اما بهبودی در پیشآگهی نهایی بینایی نشان داده نشد.
پس از بهبود کاهش بینایی و نقص میدان بینایی، برای پیشگیری از عود، DMT با همکاری متخصص مغز و اعصاب در نظر گرفته میشود.
DMTهای اصلی و اثربخشی آنها در زیر آورده شده است.
| دارو | مکانیسم اثر | روش تجویز | کاهش خطر نسبی |
|---|---|---|---|
| اینترفرون بتا | تنظیم فعالیت سلولهای T/B و ترشح سیتوکین | تزریق خودکار | RR پیشرفت ناتوانی 0.71 |
| گلاتیرامر استات | تنظیم سلولهای T تنظیمی | تزریق خودکار | RR عود 0.82 |
| ناتالیزوماب | ممانعت از ورود سلولهای التهابی به CNS | انفوزیون وریدی | RR عود 0.56 |
| فینگولیمود | تنظیم گیرنده S1P | خوراکی | RR ضایعات T2 جدید 0.65 |
| تریفلونوماید | مهار سنتز پیریمیدین | خوراکی | RR پیشرفت ناتوانی 0.76 |
| دیمتیل فومارات | کاهش استرس اکسیداتیو و التهاب | خوراکی | RR عود 0.64 |
| آلمتوزوماب | آنتیبادی مونوکلونال ضد CD52 | تزریق وریدی | RR پیشرفت ناتوانی 0.44 |
آنتیبادیهای مونوکلونال ضد CD20 (اوکرلیزوماب، ریتوکسیماب، افاتوموماب) به عنوان درمان استاندارد برای MS عودکننده تثبیت شدهاند 3).
حتی در نوریت اپتیک بدون ضایعه مغزی، ۲۵٪ موارد پس از ۱۵ سال به MS تبدیل میشوند و در صورت وجود ضایعه مغزی، ۷۸٪ موارد به MS تبدیل میشوند.
حتی در صورت عدم وجود ضایعه در MRI مغز، ۲۵٪ موارد پس از ۱۵ سال به MS مبتلا میشوند و در صورت وجود ضایعه مغزی، ۷۸٪ موارد به MS تبدیل میشوند. بیماران مبتلا به نوریت اپتیک باید با همکاری متخصص مغز و اعصاب، استفاده از DMT برای پیشگیری از عود را بررسی کنند.
MS یک بیماری خودایمنی در نظر گرفته میشود. لنفوسیتهای T میلین را به عنوان یک عامل خارجی شناسایی کرده و با فعال کردن ماکروفاژها، سیتوکینها و آنتیبادیها، میلین و آکسون را تخریب میکنند. از بین رفتن میلین باعث اختلال در هدایت ایمپالسهای الکتریکی و در نتیجه اختلال در انتقال سیگنالهای عصبی میشود.
پلاک فعال
ماکروفاژهای کفآلود: تجمع ماکروفاژهایی که میلین را فاگوسیتوز کردهاند.
نفوذ دور عروقی (perivascular cuffing): یافته مشخصه که لنفوسیتها اطراف عروق را احاطه میکنند.
ضایعات دمیلیناسیون موضعی ادماتوز: در فاز تشدید حاد دیده میشوند.
پلاک مزمن
از دست رفتن میلین: با رنگآمیزی Luxol fast blue قابل تأیید است. آکسونها حفظ میشوند اما بازسازی میلین ناقص است.
ضایعات NAWM: گلیوز منتشر، فعال شدن میکروگلیا و تخریب سد خونی-مغزی در ماده سفید به ظاهر طبیعی. همبستگی بالاتری با ناتوانی بالینی نسبت به ضایعات موضعی ماده سفید دارد.
الیگودندروسیتها مسئول بازسازی میلین در CNS هستند1). این فرآیند به سلولهای پیشساز الیگودندروسیت (OPC) بزرگسال وابسته است، اما الیگودندروسیتهای بالغ موجود نمیتوانند در بازسازی میلین مشارکت کنند1).
علل اصلی شکست بازسازی میلین به شرح زیر است1).
همچنین، آسیب ماده خاکستری قشری و زیرقشری نیز مشاهده میشود و هنگامی که ساختارهای لنفاوی فولیکول مانند سلول B در مننژ تشکیل میشود، منجر به سیر بالینی شدیدتری میشود 1).
این یک رویکرد جدید است که با مهار CD40L، تحریک همزمان سلولهای T و سلولهای ارائهدهنده آنتیژن (شامل سلولهای B) را مسدود میکند.
در کارآزمایی فاز 2 توسط Vermersch و همکاران (N Engl J Med 2024)، frexalimab اثربخشی واضحی را در برابر دارونما در پیامدهای MRI (ضایعات جدید با افزایش گادولینیوم در هفتههای 8 تا 12) نشان داد و کاهش NfL سرم، یک نشانگر زیستی آسیب بافت عصبی، نیز تأیید شد 3). برای MS پیشرونده، انتظار میرود که اثر غیرفعالسازی میکروگلیا و ماکروفاژها نیز داشته باشد و از نظر تئوری، محافظت عصبی از طریق مسدود کردن سیگنال CD40L به میکروگلیا در لبه پلاک امکانپذیر است 3).
ایجاد برتری بالینی نسبت به DMTهای مؤثر فعلی (داروهای ضد CD20) به عنوان چالش آینده در نظر گرفته میشود 3).
نشان داده شده است که فروپتوز، یک مرگ سلولی وابسته به آهن، در مرگ سلولهای عصبی در MS نقش دارد.
Tang et al. (2025) مطالعه Woo et al. (Cell, 2024) را تفسیر کرده و آبشار زیر را گزارش کردهاند: سمیت تحریکی گلوتامات → اضافه بار کلسیم → استرس شبکه آندوپلاسمی → جدا شدن STING1 از STIM1 → فعال شدن مسیر غیرکلاسیک → اتوفاژی → تجزیه اتوفاژی GPX4 (آنزیم خنثیکننده پراکسیداسیون لیپیدی) → فروپتوزیس 4). افزایش بیان STING1 در نورونها هم در نمونههای MS انسانی و هم در مدلهای موشی تأیید شده است. مهارکنندههای STING1 (C176، H151) تجزیه GPX4 وابسته به اتوفاژی را در مدلهای حیوانی کاهش داده و اثر محافظت عصبی نشان دادهاند 4).
در مرحله تحقیقاتی، محافظت عصبی از طریق غیرفعالسازی میکروگلیا و ماکروفاژها توسط مهارکننده CD40L به نام frexalimab 3) و مهار فروپتوزیس (مرگ سلولی وابسته به آهن) با مهار STING1 4) امیدوارکننده به نظر میرسند. هر دوی این روشها در حال حاضر در مرحله کارآزمایی بالینی یا تحقیقاتی هستند و درمان استاندارد محسوب نمیشوند.