نوریت اپتیک بیماری است که در آن به دلایلی عصب بینایی ملتهب شده و عملکرد بینایی کاهش مییابد. پس از رد نوروپاتیهای عفونی و غیرعفونی (سمی، ارثی، فشاری)، اکثر موارد ایدیوپاتیک هستند که علت آن خودایمنی فرض میشود.
سن شایع ۱۵ تا ۴۵ سال، بیشتر در زنان، و به صورت نوروپاتی حاد یک طرفه بینایی تظاهر میکند. اختلال بینایی طی چند روز تا دو هفته پیشرفت میکند و سپس طی پنج هفته تمایل به بهبود نشان میدهد.
بروز سالانه در ژاپن ۱.۶ نفر در هر ۱۰۰٬۰۰۰ بزرگسال است. سن شایع ۱۵ تا ۴۵ سال و حدود ۷۰٪ زنان هستند. شیوع جهانی حدود ۱ تا ۵ نفر در هر ۱۰۰٬۰۰۰ نفر تخمین زده میشود.
ارتباط بسیار نزدیکی با MS دارد و احتمال تجمعی انتقال به MS طی 15 سال پس از شروع نوریت اپتیک ایدیوپاتیک 50% است. اگر در MRI مغز در زمان اولین حمله ضایعات دمیلینه وجود نداشته باشد، احتمال انتقال تنها 25% است، اما اگر یک یا چند ضایعه وجود داشته باشد به 78% میرسد.
در کودکان، سن شایع بروز 9 تا 10 سال است و هرچه سن کمتر باشد، درگیری دوطرفه و کاهش شدیدتر بینایی دیده میشود. این امر به ویژه در کودکان زیر 5 سال بارز است. میزان انتقال به MS کودکان توسط آداچی و همکاران حدود 30% و توسط میزوتا و همکاران 16% گزارش شده است.
بروز سالانه MOGAD به ازای هر یک میلیون نفر 1.6 تا 4.8 تخمین زده میشود. 1)
Qآیا پس از ابتلا به نوریت اپتیک در آینده به مولتیپل اسکلروزیس مبتلا میشوم؟
A
حدود 50% از موارد نوریت اپتیک ایدیوپاتیک طی 15 سال به MS تبدیل میشوند. با این حال، اگر در MRI مغز در زمان اولین حمله ضایعات دمیلینه وجود نداشته باشد، احتمال انتقال تنها 25% است. پس از شروع بیماری، ارزیابی خطر با MRI مغز و پیگیری همراه با همکاری متخصص مغز و اعصاب مهم است.
Wikimedia Commons. File:Optic_Neuritis.png. License: CC BY-SA.
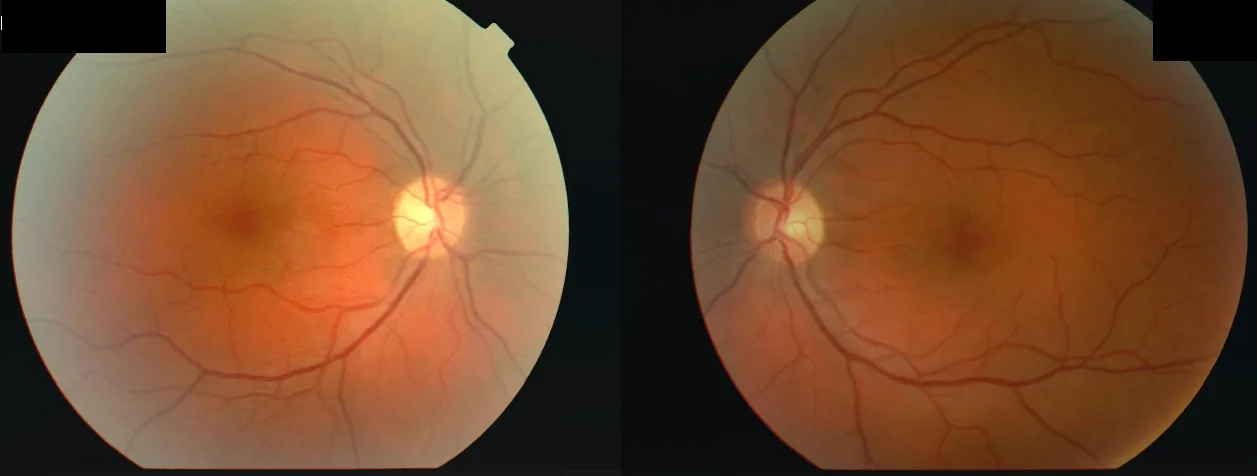
تصاویر فوندوس هر دو چشم یک زن 34 ساله که پس از یک بیماری تبدار، ابتدا در چشم راست (OD 2/60) و سپس در چشم چپ (OS 6/24) دچار کاهش بینایی شد و اسکوتوم مرکزی دوطرفه و نقص نسبی آوران مردمک (RAPD) در چشم راست داشت. این تصویر مربوط به «اسکوتوم مرکزی» است که در بخش «2. علائم اصلی و یافتههای بالینی» بحث شده است.
نقص نسبی آوران مردمک (RAPD): مهمترین یافته. در موارد یکطرفه یا با تفاوت بین دو چشم مثبت میشود. در نوریت بینایی حتی با اختلال عملکرد خفیف نیز غیرطبیعی است. قبل از گشاد کردن مردمک با لنز جلویی و لامپ شکافی بررسی شود
شدت اختلال بینایی: در مطالعه ONTT، 10% موارد حدت بینایی 1.0 یا بهتر، 25% بین 0.5 تا 1.0، 29% بین 0.1 تا 0.5 و 36% کمتر از 0.1 گزارش شده است
یافتههای فوندوس: در نوریت بینایی قدامی، پرخونی و تورم دیسک بینایی دیده میشود و در آنژیوگرافی فلورسئین هیپرفلورسانس دارد. در نوریت رتروبولبار، در مراحل اولیه طبیعی است و پس از ۴ تا ۶ هفته رنگپریدگی ایجاد میشود
افتالمپلژی بینهستهای: در موارد همراه با MS به دلیل دمیلیناسیون فاسیکولوس طولی داخلی ظاهر میشود
تشکیل غلاف عروقی شبکیه و یووئیت محیطی: در ۵ تا ۱۰٪ بیماران MS
در نوریت اپتیک کودکان، برخلاف نوریت رتروبولبار که در بزرگسالان دیده میشود، اغلب پاپیلیت معمولی همراه با قرمزی و تورم دیسک بینایی مشاهده میشود. محل التهاب فقط تورم دیسک نیست، بلکه کل طول عصب بینایی را درگیر میکند. درگیری دوطرفه در ۵۰-۷۵٪ و نوع نوریت اپتیک قدامی در ۵۰-۷۵٪ موارد رخ میدهد.
Qعلامت اوهتوف چیست؟
A
علامت اوهتوف پدیدهای است که در آن با افزایش دمای بدن، اختلال بینایی گذرا رخ میدهد. بیمار پس از حمام یا ورزش، کاهش بینایی یا تیرگی دید را تجربه میکند. این علامت پس از چند دقیقه ظاهر شده و ظرف یک ساعت از بین میرود. اگرچه در نوریت اپتیک همراه با MS شناخته شده است، اما در سایر نوروپاتیهای بینایی نیز گزارش شده و اختصاصی MS نیست.
Qنوریت اپتیک با آنتیبادی ضد AQP4 مثبت چه تفاوتی با نوریت اپتیک معمولی دارد؟
A
نوریت اپتیک با آنتیبادی ضد AQP4 مثبت یک نوع مقاوم به درمان است که حدود ۱۰٪ از نوریتهای اپتیک ایدیوپاتیک را تشکیل میدهد. در مقایسه با نوریت اپتیک معمولی، بینایی در زمان شروع بدتر است، پاسخ به درمان با استروئید ضعیفتر است و عود بیشتری دارد. همچنین تمایل بیشتری به درگیری دوطرفه دارد. برای تشخیص، اندازهگیری آنتیبادی ضد AQP4 سرم ضروری است.
در نوع ایدیوپاتیک که مکانیسم خودایمنی مطرح است، سلولهای التهابی مانند میکروگلیا به داخل عصب بینایی نفوذ کرده و باعث التهاب میشوند. التهاب مکرر منجر به آسیب تجمعی به رشتههای عصبی و در نهایت آتروفی عصب بینایی میشود.
فرضیهای مطرح است که عفونت ویروسی باعث تحریک پاسخ خودایمنی میشود و در مایع مغزی-نخاعی برخی بیماران آنتیبادی علیه ویروسهای سرخک، آبله مرغان و آنفلوانزا یافت میشود. در کودکان، ارتباط با ADEM (آنسفالومیلیت حاد منتشر) همراه با تب و سردرد شناخته شده است.
جنسیت: در زنان شایعتر است (نسبت زن به مرد حدود ۱:۲ تا ۱:۳)
سن: شایع در سنین ۲۰ تا ۴۵ سال
علائم پیشدرآمد: ممکن است یک بیماری شبهآنفلوانزا پیش از آن رخ دهد
بیماران MS: تا ۷۵٪ حداقل یک بار در طول زندگی خود نوریت اپتیک را تجربه میکنند. در کالبدشکافی، تا ۹۰٪ ضایعات عصب بینایی دارند
واکسیناسیون و عفونت به عنوان محرکهای شروع MOGAD گزارش شدهاند و مکانیسمهای شکست تحمل ایمنی مانند فعالسازی bystander فرض میشود. 1)
گزارشهایی از شروع نوریت اپتیک پس از عفونت COVID-19 یا واکسیناسیون وجود دارد. میانه زمان از واکسیناسیون تا شروع نوریت اپتیک ۱۸ روز بود و در ۱۴ مورد از ۵۵ مورد MOG-IgG مثبت بود. 11)
ضخامت لایه فیبرهای عصبی شبکیه اطراف پاپی (pRNFL) اندازهگیری میشود. در مرحله حاد، ضخیمشدن pRNFL (میانه MOG-ON 164 میکرومتر در مقابل MS-ON 103 میکرومتر) مشاهده میشود. در مرحله مزمن، نازکشدن pRNFL رخ میدهد و تعداد عودها با کاهش pRNFL همبستگی دارد. در مقادیر pRNFL کمتر از آستانه 50 میکرومتر، mean deviation میدان بینایی به طور معنیداری بدتر میشود.1)
در صورت سیر غیرمعمول، موارد زیر اندازهگیری میشوند.
آنتیAQP4: تحت پوشش بیمه از سال 2013. در صورت مقاومت به استروئید یا عود مکرر، ارزیابی زودهنگام مهم است
آنتیMOG (MOG-IgG): آزمایش سرم با روش سلول زنده (live cell-based assay) توصیه میشود. روش CBA ثابت حساسیت و ویژگی کمتری دارد
بر اساس معیارهای تشخیصی بینالمللی MOGAD 2023، تشخیص قطعی با CBA مثبت + فنوتیپ بالینی اصلی + حذف تشخیصهای جایگزین انجام میشود. در تیتر پایین، حداقل یک ویژگی بالینی/تصویربرداری حمایتکننده لازم است2)
اعتبارسنجی این معیارها: حساسیت 96.5%، ویژگی 98.9%، دقت 98.5%3)
احتمال اینکه نوریت اپتیک علامت اولیه MS باشد ارزیابی میشود. تشخیص با اثبات انتشار زمانی و مکانی ضایعات دمیلینه التهابی در سیستم عصبی مرکزی و حذف سایر بیماریها انجام میشود. وجود ضایعات T2 با سیگنال بالا در دو یا بیش از دو ناحیه از چهار ناحیه (پریبطنی، زیرقشری، اینفراتنتوریال، نخاع) انتشار مکانی را اثبات میکند.
گزارش شده است که مواردی که به عنوان نوریت رتروبولبار تشخیص داده شدهاند، در واقع نوروپاتی فشاری ناشی از لنفوم اربیت بودهاند. بنابراین در صورت وجود ویژگیهای غیرمعمول، نباید به راحتی استروئید تجویز کرد و باید بررسی کامل انجام شود. 9)
معمولاً FA (آنژیوگرافی فلورسئین)، CFF و میدان بینایی پویا که در بزرگسالان انجام میشود، برای کودکان مناسب نیست. تشخیص بر اساس یافتههای فوندوسکوپی و MRI مغز انجام میشود.
Qنکات کلیدی در MRI برای تمایز نوریت اپتیک تیپیک و آتیپیک چیست؟
A
در نوریت اپتیک تیپیک، تنها یک بخش کوتاه از عصب بینایی افزایش کنتراست نشان میدهد. در مقابل، در نوریت اپتیک آتیپیک، افزایش کنتراست طولانی (بیش از نیمی از طول عصب)، گسترش به سمت عقب (کیاسم و دستگاه بینایی) و افزایش کنتراست غلاف عصب بینایی دیده میشود. این یافتههای MRI مستقیماً به تشخیص افتراقی بیماری زمینهای کمک میکنند.
پیشآگهی بینایی در نوریت اپتیک ایدیوپاتیک خوب است. در بیش از 90٪ موارد نوریت اپتیک، بینایی با مشاهده یا تجویز سیستمیک استروئید بهبود مییابد. بر اساس نتایج طولانیمدت ONTT، در 93٪ موارد یک سال پس از شروع، حدت بینایی 0.5 یا بهتر و در بیش از 70٪ موارد 1.0 یا بهتر است.
در نوریت اپتیک ایدیوپاتیک با حدت بینایی اصلاحشده نسبتاً خوب، میتوان با مکوبالامین خوراکی 1500 میکروگرم در روز (خارج از برچسب) پیگیری کرد.
نوریت اپتیک تیپیک
خط اول: پالس استروئیدی (متیلپردنیزولون 1000 میلیگرم در روز انفوزیون وریدی به مدت 3 روز). خارج از برچسب.
پس از پالس: شروع پردنیزولون خوراکی با دوز 0.5 میلیگرم/کیلوگرم/روز و سپس کاهش تدریجی 5 تا 10 میلیگرم هر 3 تا 4 روز.
در صورت عدم پاسخ به پالس اول: پالس دوم پس از 4 تا 5 روز انجام میشود.
اثر: مدت زمان بهبودی را کوتاه میکند، اما تفاوت معنیداری در حدت بینایی نهایی یک سال پس از شروع وجود ندارد.
اندیکاسیون فعال: درگیری دوطرفه، اختلال شدید عملکرد بینایی، تنها چشم عملکردی، موارد عودکننده، وجود پلاکهای دمیلیناسیون در MRI، و زمانی که بیمار به شدت خواهان بهبودی زودهنگام است.
ON با آنتیبادی ضد AQP4 مثبت
فاز حاد: پالس استروئیدی (خط اول درمان).
در صورت عدم پاسخ: تکرار پالس پس از 3-4 روز → در صورت عدم پاسخ → پلاسمافرز در نظر گرفته شود.
پلاسمافرز: پلاسمافرز ساده > پلاسمافرز با فیلتر غشایی دوگانه > ایمونوجذب (به ترتیب اثربخشی). یک دوره 5-6 جلسه.
درمان با پردنیزولون خوراکی با دوز استاندارد (1 میلیگرم/کیلوگرم/روز) به تنهایی در مطالعه ONTT نسبت به دارونما یا استروئید داخل وریدی میزان عود بالاتری داشت، بنابراین توصیه نمیشود.
در موارد همراه با MS، پس از بهبود بینایی، درمان برای پیشگیری از عود در نظر گرفته میشود. با همکاری متخصص مغز و اعصاب، داروهای تعدیلکننده بیماری (مانند اینترفرون بتا، گلاتیرامر استات، فینگولیمود، ناتالیزوماب) بررسی میشوند.
Qآیا درمان با استروئید بر بهبود نهایی بینایی تأثیر میگذارد؟
A
نتایج ONTT نشان داد که پالس درمانی با استروئید سرعت بهبود را افزایش میدهد، اما در دید نهایی یک سال پس از شروع تفاوت معنیداری وجود نداشت. در نوریت اپتیک ایدیوپاتیک، حتی بدون درمان، ۹۳٪ موارد به دید ۰.۵ یا بهتر میرسند. با این حال، موارد مثبت آنتیAQP4 به استروئید مقاوم هستند و نیاز به درمان تهاجمی اولیه از جمله پلاسمافرز دارند.
Qدرمان سرکوبکننده ایمنی تا چه مدت باید ادامه یابد؟
A
از آنجایی که CRION بیماری است که با قطع درمان عود میکند، اغلب نیاز به ادامه طولانیمدت درمان سرکوبکننده ایمنی حتی پس از بهبود علائم وجود دارد. پس از تعیین حداقل دوز مؤثر استروئید، داروهای سرکوبکننده ایمنی غیراستروئیدی نیز اضافه میشوند و رژیم درمانی به صورت فردی طراحی میشود.
در موارد ایدیوپاتیک که مکانیسم خودایمنی مطرح است، سلولهای التهابی مانند میکروگلیا به عصب بینایی نفوذ کرده و باعث التهاب میشوند.
تخریب میلین با واسطه ایمنی: غلاف میلین عصب بینایی توسط مکانیسم خودایمنی مورد حمله قرار میگیرد. هدایت جهشی مختل شده و اختلال در هدایت ایمپالس آکسونی رخ میدهد
دژنراسیون آکسونی: پس از تخریب میلین، آکسونهای سلولهای گانگلیونی شبکیه شروع به دژنره شدن میکنند
فاگوسیتوز توسط ماکروفاژها: ماکروفاژها میلین باقیمانده را حذف میکنند
گلیوز: آستروسیتها تکثیر یافته و اسکار گلیال تشکیل میدهند. «اسکلروز» در مولتیپل اسکلروزیس از این پدیده ناشی میشود
التهاب مکرر عصب بینایی منجر به آسیب تجمعی به رشتههای عصبی و در نهایت آتروفی عصب بینایی میشود.
آنتیAQP4 با کمپلمان ترکیب شده و به آستروسیتها، سلولهای گلیال در عصب بینایی، حمله میکند و باعث بروز بیماری میشود. آستروسیتهای عصب بینایی و کیاسمای بینایی AQP4 زیادی بیان میکنند و هدف آسانی هستند.
MOG-IgG (عمدتاً زیرکلاس IgG1) MOG را روی سطح میلین CNS هدف قرار میدهد. فعال شدن مسیر کمپلمان و فاگوسیتوز سلولی وابسته به آنتیبادی در دمیلیناسیون نقش دارند. با این حال، فعال شدن کمپلمان در مقایسه با AQP4-IgG ضعیفتر است. در MOGAD، سلولهای T CD4+ و ماکروفاژها غالب هستند، در حالی که در MS، سلولهای T CD8+ غالب هستند. 1)4)
اگرچه MOG در شبکیه بیان نمیشود، در MOG-ON آسیب به سلولهای گانگلیونی شبکیه رخ میدهد. سمیت گلوتامات و شکنندگی سد خونی-مغزی در سر عصب بینایی به عنوان مکانیسمهای این پدیده مطرح شدهاند. 1)
همچنین مکانیسم افزایش نفوذپذیری سد خونی-مغزی توسط IL-6 و تسریع تمایز پلاسمابلاستها مورد توجه قرار گرفته است.
CRION در ابتدا به عنوان سندرمی شامل نوریت اپتیک حساس به استروئید و عودکننده مطرح شد. 5) آزمایش آنتیبادی در کوهورتهای بعدی CRION نشان داد که حداکثر 22% AQP4-IgG مثبت و حداکثر 25% MOG-IgG مثبت هستند. 6)7)8) بنابراین، CRION یک تشخیص سندرمی است که شامل گروههای اتیولوژیک ناهمگن از جمله نوریت اپتیک مرتبط با آنتیبادی MOG و نوریت اپتیک مرتبط با آنتیبادی AQP4 میشود.
فرضیهای مطرح شده است که عفونت ویروسی باعث ایجاد پاسخ خودایمنی میشود. گزارشهایی از تشخیص DNA ویروس واریسلا-زوستر و ویروس هرپس سیمپلکس در مایع مغزی-نخاعی برخی از بیماران ON وجود دارد. در MOGAD، تصور میشود که واکسیناسیون یا عفونت باعث شکست تحمل ایمنی (فعال شدن bystander و تقلید مولکولی) میشود. 1)
فاز 3 کارآزمایی بالینی تصادفیشده برای ساترالیزومب (NCT05271409) و روزانولیکسیزومب (NCT05063162) در حال انجام است. استفاده خارج از برچسب توسیلیزومب (آنتیبادی گیرنده IL-6) اثر پیشگیری از عود تا 29 ماه را نشان داده است.1)
مواردی از نوریت اپتیک دمیلینهکننده پس از عفونت COVID-19 گزارش شده است. جاسی و همکاران (2022) مواردی از نوریت اپتیک در دوره نقاهت عفونت را گزارش کردند که همه با پالس استروئیدی بهبود بینایی یافتند.10)
گزارشهایی از بروز نوریت اپتیک پس از واکسیناسیون COVID-19 جمعآوری شده است که میانه زمان بروز 18 روز پس از واکسیناسیون است. از 55 مورد، 14 مورد MOG-IgG مثبت بودند و هیچ موردی AQP4-IgG مثبت نداشت.11) همچنین مواردی از نوریت اپتیک مرتبط با آنتیبادی MOG پس از عفونت SARS-CoV-2 گزارش شده است.12)
با گسترش آزمایشهای آنتیبادی MOG-IgG و AQP4-IgG، برخی از مواردی که قبلاً CRION تشخیص داده میشدند، اکنون به MOGAD یا NMOSD طبقهبندی میشوند. انتظار میرود بازتعریف مفاهیم بیماری بر اساس پروفایل آنتیبادی در آینده پیشرفت کند.
Jeyakumar N, Lerch M, Dale RC, Ramanathan S. MOG antibody-associated optic neuritis. Eye (London, England). 2024;38(12):2289-2301. doi:10.1038/s41433-024-03108-y. PMID:38783085; PMCID:PMC11306565.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. The Lancet. Neurology. 2023;22(3):268-282. doi:10.1016/S1474-4422(22)00431-8. PMID:36706773.
Varley JA, Champsas D, Prossor T, et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology. 2024.
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2019;90(9):1021-1026. doi:10.1136/jnnp-2019-320493. PMID:31118222.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. Journal of neuroinflammation. 2018;15(1):302. doi:10.1186/s12974-018-1335-x. PMID:30382857; PMCID:PMC6208174.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. Journal of neurology, neurosurgery, and psychiatry. 2010;81(1):109-11. doi:10.1136/jnnp.2008.146894. PMID:20019228.
McElhinney K, Rhatigan M, Tsvetanova Z, O’Keane C, Logan P. Beware the Retrobulbar Optic Neuritis Diagnosis. Case reports in ophthalmology. 2022;13(2):453-458. doi:10.1159/000524685. PMID:35950025; PMCID:PMC9247490.
Jossy A, Jacob N, Sarkar S, Gokhale T, Kaliaperumal S, Deb AK. COVID-19-associated optic neuritis - A case series and review of literature. Indian journal of ophthalmology. 2022;70(1):310-316. doi:10.4103/ijo.IJO_2235_21. PMID:34937266; PMCID:PMC8917537.
Bhatti MT, Gilbert AL, Watson G, et al. Shot in the dark. Surv Ophthalmol. 2023;68:821-829.
Francesca Bosello, Damiano Marastoni, Francesca Benedetta Pizzini, Chiara Zaffalon, Andrea Zuliani, Giulia Turri, Sara Mariotto, Erika Bonacci, et al. Atypical myelin oligodendrocyte glycoprotein antibody–associated optic neuritis and acute demyelinating polyneuropathy after SARS-CoV-2 infection: Case report and literature review. Journal of Neuroimmunology. 2023;375:578011. doi:10.1016/j.jneuroim.2022.578011.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. Journal of neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0. PMID:27793206; PMCID:PMC5086042.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. Journal of neurology, neurosurgery, and psychiatry. 2018;89(2):127-137. doi:10.1136/jnnp-2017-316880. PMID:29142145; PMCID:PMC5800335.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. MOG antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.