La neuritis óptica es una enfermedad en la que el nervio óptico se inflama por alguna causa, lo que lleva a una disminución de la función visual. Después de excluir las neuropatías ópticas infecciosas y no infecciosas (tóxicas, hereditarias, compresivas), la mayoría son idiopáticas, consideradas causadas por mecanismos autoinmunes.
La edad de aparición más frecuente es entre 15 y 45 años, más común en mujeres, y se presenta como neuropatía óptica aguda monocular. La disfunción visual progresa durante varios días a 2 semanas, luego muestra una tendencia a recuperarse dentro de las 5 semanas.
Neuritis óptica anterior (papilitis): Acompañada de edema del disco óptico. Se observa en aproximadamente el 50% en Japón (más alto que el 35% en países occidentales).
Neuritis óptica retrobulbar: El fondo de ojo parece normal en la fase aguda. La palidez del disco óptico aparece después de 4 a 6 semanas.
La incidencia anual en Japón es de 1,6 por cada 100.000 adultos. La edad pico es de 15 a 45 años, y las mujeres representan aproximadamente el 70%. La prevalencia mundial se estima en 1 a 5 por cada 100.000 personas.
La asociación con la EM es muy fuerte, con una probabilidad acumulada de conversión a EM del 50% a los 15 años del inicio de la neuritis óptica idiopática. Si no hay lesiones desmielinizantes en la RM cerebral al inicio, la tasa de conversión se mantiene en el 25%, pero si hay una o más lesiones, alcanza el 78%.
En niños, la edad pico de aparición es de 9 a 10 años, y cuanto más pequeño es el niño, más probable es que presente afectación bilateral y pérdida visual grave, especialmente en menores de 5 años. La tasa de conversión a EM pediátrica se ha reportado como aproximadamente 30% por Adachi et al. y 16% por Mizota et al.
La incidencia anual de MOGAD se estima en 1.6–4.8 por millón de personas. 1)
QSi desarrollo neuritis óptica, ¿tendré esclerosis múltiple en el futuro?
A
Aproximadamente el 50% de los casos de neuritis óptica idiopática se convierten en EM en 15 años. Sin embargo, si no hay lesiones desmielinizantes en la RM cerebral al inicio, la tasa de conversión se mantiene en el 25%. Después del inicio, es importante realizar una evaluación de riesgo mediante RM cerebral y seguimiento en colaboración con un neurólogo.
Wikimedia Commons. File:Optic_Neuritis.png. License: CC BY-SA.
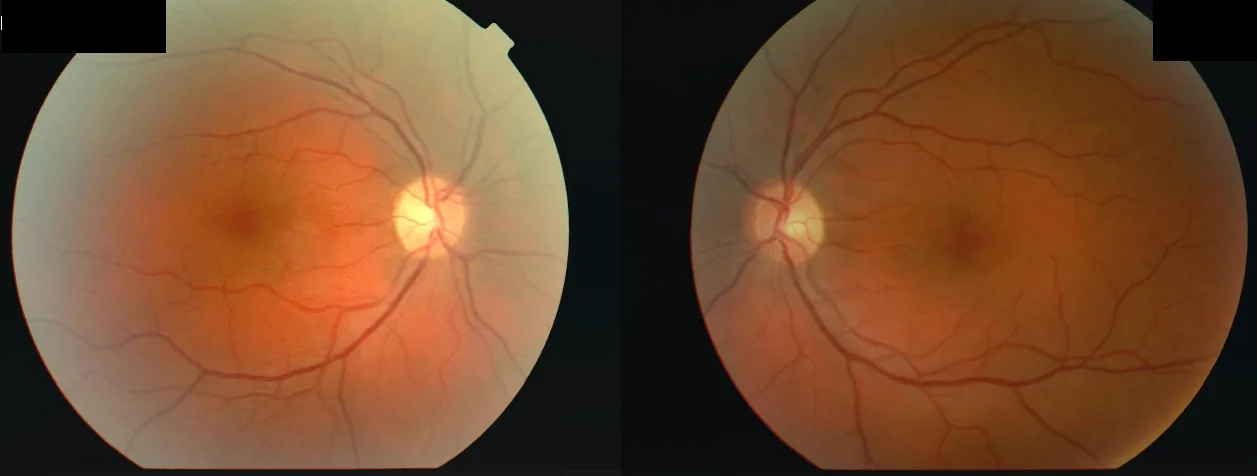
Fotografías de fondo de ojo de una mujer de 34 años que desarrolló disminución de la visión en el ojo derecho (OD 2/60) seguido del ojo izquierdo (OS 6/24) después de una enfermedad febril, con escotomas centrales en ambos ojos y un defecto pupilar aferente relativo (RAPD) en el ojo derecho. Esto corresponde al “escotoma central” discutido en la sección “2. Síntomas principales y hallazgos clínicos”.
Se presenta con un patrón de inicio agudo, y los siguientes síntomas son característicos:
Pérdida visual aguda unilateral: Progresa durante varios días hasta aproximadamente 2 semanas. La función visual es peor alrededor de 1 semana después del inicio.
Dolor ocular / dolor con el movimiento ocular: Se observa en aproximadamente el 50–60% de los casos en Japón. Puede aparecer unos días antes de la pérdida visual.
Defectos del campo visual: Son comunes el escotoma central o el escotoma centrocecal, pero también puede ocurrir hemianopsia altitudinal. Los defectos difusos también son frecuentes.
Anomalía de la visión cromática: La disminución de la saturación del color rojo es característica.
Fenómeno de Uhthoff: Pérdida visual transitoria o visión borrosa después de un aumento de la temperatura corporal (p. ej., baño caliente, ejercicio). Aparece en minutos y se resuelve en 1 hora.
Fosfenos (phosphenes): Percepción de destellos de luz
Fenómeno de Pulfrich: Alteración de la percepción de profundidad de objetos en movimiento
Defecto pupilar aferente relativo (RAPD): Hallazgo más importante. Positivo cuando es unilateral o asimétrico. En la neuritis óptica, puede ser anormal incluso con disfunción leve. Verificar con lente de condensación y lámpara de hendidura antes de la dilatación.
Grado de deterioro visual: En ONTT, agudeza visual ≥1.0 en 10%, 0.5–1.0 en 25%, 0.1–0.5 en 29%, y peor en 36%.
Hallazgos de fondo de ojo: En neuritis óptica anterior, se observa hiperemia e hinchazón del disco óptico, con hiperfluorescencia en angiografía fluoresceínica. En neuritis óptica retrobulbar, el fondo es normal al inicio, con palidez después de 4–6 semanas.
Oftalmoplejía internuclear: Aparece en pacientes con EM debido a desmielinización del fascículo longitudinal medial.
Vaina vascular retiniana y uveítis periférica: Presente en 5–10% de pacientes con EM.
En la neuritis óptica pediátrica, es más frecuente la papilitis típica con enrojecimiento e hinchazón del disco óptico que la neuritis óptica retrobulbar observada en adultos. La inflamación se extiende a lo largo de todo el nervio óptico, no solo al disco. La afectación bilateral ocurre en el 50–75% de los casos, y la forma de neuritis óptica anterior representa el 50–75%.
Q¿Qué es el signo de Uhthoff?
A
El signo de Uhthoff es un fenómeno de alteración visual transitoria asociado con un aumento de la temperatura corporal. Los pacientes notan disminución de la visión o visión borrosa después de bañarse o hacer ejercicio. Aparece en minutos y se resuelve en una hora. Se conoce en la neuritis óptica asociada a EM, pero también se ha informado en otras neuropatías ópticas y no es específico de la EM.
Q¿En qué se diferencia la neuritis óptica con anticuerpos anti-AQP4 positivos de la neuritis óptica típica?
A
La neuritis óptica con anticuerpos anti-AQP4 positivos es un subtipo refractario que representa aproximadamente el 10% de las neuritis ópticas idiopáticas. En comparación con la neuritis óptica típica, la agudeza visual al inicio es peor, la respuesta al tratamiento con esteroides es deficiente y las recurrencias son más frecuentes. También tiende a volverse bilateral. La medición de anticuerpos anti-AQP4 en suero es esencial para el diagnóstico.
En los casos idiopáticos con sospecha de mecanismo autoinmune, células inflamatorias como la microglía infiltran el nervio óptico y causan inflamación. La inflamación repetida provoca daño acumulativo a las fibras nerviosas y progresión a atrofia óptica.
Se ha propuesto la hipótesis de que una infección viral desencadena una reacción autoinmune, y se detectan anticuerpos virales contra sarampión, varicela, influenza, etc., en el líquido cefalorraquídeo de algunos pacientes. En niños, se conoce una asociación con la encefalomielitis aguda diseminada (ADEM) acompañada de fiebre y dolor de cabeza.
Sexo: Más común en mujeres (proporción hombre:mujer aproximadamente 1:2 a 1:3)
Edad: Más frecuente entre los 20 y 45 años
Síntomas prodrómicos: Puede estar precedido por una enfermedad similar a la influenza
Pacientes con EM: Hasta el 75% experimenta al menos un episodio de ON en su vida. En autopsias, hasta el 90% presenta lesiones del nervio óptico.
Se ha informado que la vacunación y la infección desencadenan la aparición de MOGAD, y se sospechan mecanismos como la activación de espectadores que conducen a la ruptura de la tolerancia inmunológica. 1)
Hay informes de ON después de la infección por COVID-19 o la vacunación. La mediana de tiempo desde la vacunación hasta la aparición de ON fue de 18 días, y 14 de 55 casos fueron MOG-IgG positivos. 11)
La neuritis óptica es un diagnóstico clínico. Se diagnostica basándose en la combinación de pérdida visual aguda unilateral, dolor con el movimiento ocular, RAPD positivo y defectos del campo visual.
RMN orbitaria: STIR con supresión grasa y T1 con contraste en plano coronal. Evaluar aumento de tamaño, hiperintensidad y realce del nervio óptico.
RMN cerebral FLAIR: Evaluar la presencia de lesiones desmielinizantes cerebrales. Esencial para evaluar la comorbilidad con EM y el riesgo futuro de desarrollar EM.
La ON típica y la atípica difieren en los hallazgos de RMN de la siguiente manera.
Característica
ON típica (relacionada con EM)
ON atípica
Patrón de realce
Realce de segmento corto
Realce de segmento largo (≥1/2)
Ubicación de la lesión
Anterior (nervio óptico retrobulbar)
Se extiende posteriormente (quiasma y tracto óptico)
Vaina del nervio óptico
Sin realce con contraste
Realce con contraste de la vaina del nervio óptico y la grasa orbitaria
Comparación de las características de imagen de NMOSD-ON, MOG-ON y MS-ON:
Mide el grosor de la capa de fibras nerviosas de la retina peripapilar (pRNFL). En la fase aguda se observa engrosamiento de la pRNFL (mediana MOG-ON 164 μm vs MS-ON 103 μm). En la fase crónica ocurre adelgazamiento de la pRNFL, y el número de recaídas se correlaciona con la reducción de la pRNFL. Por debajo del umbral de pRNFL de 50 μm, la desviación media del campo visual empeora significativamente. 1)
Anticuerpo anti-AQP4: Cubierto por el seguro en 2013. La evaluación temprana es importante en casos de resistencia a esteroides o recaídas recurrentes.
Anticuerpo anti-MOG (MOG-IgG): Se recomienda la prueba sérica mediante ensayo basado en células vivas. El CBA fijo tiene sensibilidad y especificidad inferiores.
Los criterios diagnósticos internacionales de MOGAD de 2023 definen el diagnóstico definitivo como CBA positivo + fenotipo clínico central + exclusión de diagnósticos alternativos. En caso de título bajo, se requiere al menos una característica clínica/de RM de apoyo 2).
Validación de estos criterios: sensibilidad 96.5%, especificidad 98.9%, precisión 98.5% 3).
Evaluar la posibilidad de que la neuritis óptica sea el síntoma inicial de EM. El diagnóstico se realiza demostrando diseminación en tiempo y espacio de lesiones inflamatorias desmielinizantes del sistema nervioso central y excluyendo otras enfermedades. Se puede demostrar diseminación en espacio si hay lesiones hiperintensas en T2 en al menos dos de cuatro áreas: periventricular, cortical/yuxtacortical, infratentorial y médula espinal.
Acompañado de fiebre, cefalea, alteración de la conciencia
Se ha informado de casos diagnosticados de neuritis óptica retrobulbar que en realidad eran neuropatía óptica compresiva debido a linfoma orbitario. Si hay características atípicas, no se deben administrar corticosteroides a la ligera y se debe realizar un examen exhaustivo. 9)
Por lo general, la FA (angiografía fluoresceínica), la CFF y la perimetría cinética realizadas en adultos no son adecuadas para niños; el diagnóstico se realiza mediante hallazgos de fondo de ojo y RMN de cabeza.
Q¿Cuáles son los puntos en la RMN para distinguir la neuritis óptica típica de la atípica?
A
En la ON típica, se observa realce de contraste solo en un segmento corto del nervio óptico. Por el contrario, la ON atípica muestra hallazgos como realce de contraste de segmento largo (más de la mitad de la longitud), extensión posterior (al quiasma óptico y tracto óptico) y realce de la vaina del nervio óptico. Estos hallazgos de RMN están directamente relacionados con el diagnóstico diferencial de la enfermedad subyacente.
El pronóstico visual de la neuritis óptica idiopática es bueno. En más del 90% de los casos de neuritis óptica, la agudeza visual mejora con observación o administración sistémica de corticosteroides. Según los resultados a largo plazo del ONTT, la agudeza visual al año del inicio es de 0.5 o mejor en el 93% y de 1.0 o mejor en más del 70%.
En la neuritis óptica idiopática con agudeza visual corregida relativamente buena, se puede observar con mecobalamina oral 1.500 μg/día (uso no indicado en ficha técnica).
ON típica
Primera línea: Terapia de pulso de corticosteroides (metilprednisolona 1.000 mg/día en infusión intravenosa durante 3 días). Uso no indicado en ficha técnica.
Después del pulso: Iniciar prednisolona oral a 0,5 mg/kg/día, luego reducir gradualmente 5–10 mg cada 3–4 días.
Si el primer pulso es ineficaz: Administrar un segundo pulso después de un intervalo de 4–5 días.
Efecto: Acorta el período de recuperación, pero no hay diferencia significativa en la agudeza visual final al año del inicio.
Indicación activa: Afectación bilateral, disfunción visual grave, único ojo funcional, casos recurrentes, presencia de placas desmielinizantes en RM, o el paciente desea firmemente una mejoría temprana.
ON con anticuerpos anti-AQP4 positivos
Fase aguda: Terapia de pulsos de esteroides (primera línea).
Si no hay respuesta: Repetir pulso a los 3–4 días → si aún no hay respuesta → considerar plasmaféresis.
Plasmaféresis: Plasmaféresis simple > plasmaféresis de doble filtración > inmunoadsorción (en orden de eficacia). Un ciclo: 5–6 sesiones.
Terapia de mantenimiento: Prednisolona 5–10 mg/día + azatioprina 50–100 mg/día.
Nota: A diferencia de la ON idiopática, es importante la prevención de recaídas con esteroides orales.
ON con anticuerpos anti-MOG positivos (MOGAD)
Fase aguda: La terapia de pulsos de esteroides es altamente efectiva. Recuperación completa 50%, recuperación parcial 44%. 13)
Tendencia a recaídas: Fuerte dependencia de esteroides; el 70% recae durante la reducción de prednisolona oral (especialmente <10 mg/día o dentro de los 2 meses posteriores a la suspensión). 14)
Terapia de mantenimiento: IVIg (≥1 g/kg cada 4 semanas reduce significativamente las recaídas). 15)Rituximab puede ser menos efectivo que para AQP4-ON.
Momento de inicio del tratamiento: Generalmente después del segundo ataque (porque >50% son monofásicos).
La monoterapia con prednisolona oral en dosis estándar (1 mg/kg/día) no se recomienda porque se asoció con una mayor tasa de recaídas en comparación con placebo o corticosteroides intravenosos en el ONTT.
Se realiza una terapia agresiva con corticosteroides. Si la respuesta al pulso de corticosteroides es deficiente, la inmunoglobulina intravenosa en dosis altas o la plasmaféresis son opciones.
En casos con EM, considere el tratamiento para la prevención de recaídas después de la mejoría visual. En colaboración con un neurólogo, considere fármacos modificadores de la enfermedad (interferón beta, acetato de glatiramer, fingolimod, natalizumab, etc.).
Q¿El tratamiento con esteroides afecta la recuperación visual final?
A
Según los resultados de ONTT, la terapia con pulsos de esteroides acorta el tiempo de recuperación, pero no afecta significativamente la agudeza visual final al año del inicio. En la neuritis óptica idiopática, el 93% de los pacientes no tratados se recupera a una agudeza visual de 0.5 o mejor. Sin embargo, los casos con anticuerpos anti-AQP4 positivos son resistentes a los esteroides y requieren tratamiento agresivo que incluya recambio plasmático temprano.
Q¿Por cuánto tiempo se debe continuar la terapia inmunosupresora?
A
Dado que CRION es una enfermedad que recae al suspender el tratamiento, a menudo es necesaria la terapia inmunosupresora a largo plazo incluso después de la remisión de los síntomas. Después de identificar la dosis mínima efectiva de esteroides, se añaden inmunosupresores no esteroideos y se diseña un régimen de tratamiento individualizado.
En los casos idiopáticos en los que se sospecha un mecanismo autoinmune, células inflamatorias como la microglía infiltran el nervio óptico y causan inflamación.
Desmielinización inmunomediada: La vaina de mielina del nervio óptico es atacada por mecanismos autoinmunes. La conducción saltatoria se vuelve imposible, lo que provoca un trastorno en la conducción del impulso axonal.
Degeneración axonal: Después de la desmielinización, los axones de las células ganglionares de la retina comienzan a degenerar.
Fagocitosis por macrófagos: Los macrófagos eliminan la mielina residual.
Gliosis: Los astrocitos proliferan y forman cicatrices gliales. El término “esclerosis” en la esclerosis múltiple se origina de este proceso.
La inflamación repetida del nervio óptico causa daño acumulativo a las fibras nerviosas, lo que lleva a la atrofia óptica.
Mecanismo de la neuritis óptica con anticuerpos anti-AQP4 positivos
Los anticuerpos anti-AQP4 se unen al complemento y atacan a los astrocitos, las células gliales del nervio óptico, lo que desencadena la enfermedad. Los astrocitos del nervio óptico y del quiasma óptico expresan altos niveles de AQP4, lo que los convierte en blancos susceptibles.
Mecanismo de la ON positiva para anticuerpos anti-MOG
MOG-IgG (principalmente subclase IgG1) se dirige a MOG en la superficie de la mielina del SNC. La activación de la vía del complemento y la fagocitosis celular dependiente de anticuerpos participan en la desmielinización. Sin embargo, la activación del complemento es más débil en comparación con AQP4-IgG. En MOGAD, predominan los linfocitos T CD4 positivos y los macrófagos, mientras que en la EM predominan los linfocitos T CD8 positivos. 1)4)
A pesar de que MOG no se expresa en la retina, la MOG-ON causa daño a las células ganglionares de la retina. Se han propuesto como mecanismos la toxicidad por glutamato y la vulnerabilidad de la barrera hematoencefálica en la cabeza del nervio óptico. 1)
También se está prestando atención al mecanismo por el cual la IL-6 aumenta la permeabilidad de la barrera hematoencefálica y promueve la diferenciación de plasmoblastos.
CRION fue propuesto originalmente como un síndrome que abarca la neuritis óptica sensible a esteroides y recurrente. 5) Las pruebas de anticuerpos posteriores en cohortes de CRION revelaron que hasta el 22% son positivos para AQP4-IgG y hasta el 25% son positivos para MOG-IgG. 6)7)8) Por lo tanto, CRION se considera un diagnóstico sindrómico que engloba etiologías heterogéneas, incluyendo la neuritis óptica asociada a anticuerpos MOG y la neuritis óptica asociada a anticuerpos AQP4.
Se ha propuesto la hipótesis de que la infección viral desencadena reacciones autoinmunes. Hay informes de detección de ADN del virus varicela-zóster y del virus del herpes simple en el líquido cefalorraquídeo de algunos pacientes con ON. En MOGAD, se asume una ruptura de la tolerancia inmune debido a la vacunación o infección (activación por espectador, mimetismo molecular). 1)
7. Investigación más reciente y perspectivas futuras
Han surgido agentes biológicos con evidencia de Nivel 1 para NMOSD positivo para AQP4. 4)
Eculizumab: Inhibidor de C5. El ensayo de fase 3 mostró una reducción del 94% en el riesgo de recaída
Ravulizumab: Inhibidor de C5 (versión mejorada de eculizumab). El ensayo de fase 3 mostró una reducción del 98.6% en el riesgo de recaída
Inebilizumab: fármaco depletor de células B dirigido a CD19. Reduce el riesgo de recaída en un 77% en NMOSD positivo para AQP4.
Satralizumab: inhibidor del receptor de IL-6. Reduce el riesgo de recaída en un 74-79% en NMOSD positivo para AQP4. Puede administrarse por vía subcutánea en el hogar.
Ambos tienen evidencia establecida para NMOSD positivo para AQP4, pero su indicación para MOGAD aún no está establecida.
Están en curso los ensayos clínicos de fase 3 de satralizumab (NCT05271409) y rozanolixizumab (NCT05063162). El uso fuera de indicación de tocilizumab (anticuerpo contra el receptor de IL-6) ha mostrado prevención de recaídas hasta por 29 meses. 1)
Se han reportado casos de neuritis óptica desmielinizante después de la infección por COVID-19. Jossy et al. (2022) reportaron casos de neuritis óptica durante la fase de recuperación de la infección, y todos los pacientes recuperaron la visión con terapia de pulsos de esteroides. 10)
Se han acumulado reportes de inicio de neuritis óptica después de la vacunación contra COVID-19, con una mediana de 18 días desde la vacunación hasta el inicio. De 55 casos, 14 fueron positivos para MOG-IgG y ninguno fue positivo para AQP4-IgG. 11) También se han reportado casos de neuritis óptica asociada a anticuerpos MOG después de la infección por SARS-CoV-2. 12)
Con la difusión de las pruebas de anticuerpos MOG-IgG y AQP4-IgG, algunos casos previamente diagnosticados como CRION están siendo reclasificados como MOGAD o NMOSD. Se espera que en el futuro avance la redefinición del concepto de enfermedad basada en el perfil de anticuerpos.
Jeyakumar N, Lerch M, Dale RC, Ramanathan S. MOG antibody-associated optic neuritis. Eye (London, England). 2024;38(12):2289-2301. doi:10.1038/s41433-024-03108-y. PMID:38783085; PMCID:PMC11306565.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. The Lancet. Neurology. 2023;22(3):268-282. doi:10.1016/S1474-4422(22)00431-8. PMID:36706773.
Varley JA, Champsas D, Prossor T, et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology. 2024.
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCID:PMC10658081.
Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2019;90(9):1021-1026. doi:10.1136/jnnp-2019-320493. PMID:31118222.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. Journal of neuroinflammation. 2018;15(1):302. doi:10.1186/s12974-018-1335-x. PMID:30382857; PMCID:PMC6208174.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. Journal of neurology, neurosurgery, and psychiatry. 2010;81(1):109-11. doi:10.1136/jnnp.2008.146894. PMID:20019228.
McElhinney K, Rhatigan M, Tsvetanova Z, O’Keane C, Logan P. Beware the Retrobulbar Optic Neuritis Diagnosis. Case reports in ophthalmology. 2022;13(2):453-458. doi:10.1159/000524685. PMID:35950025; PMCID:PMC9247490.
Jossy A, Jacob N, Sarkar S, Gokhale T, Kaliaperumal S, Deb AK. COVID-19-associated optic neuritis - A case series and review of literature. Indian journal of ophthalmology. 2022;70(1):310-316. doi:10.4103/ijo.IJO_2235_21. PMID:34937266; PMCID:PMC8917537.
Bhatti MT, Gilbert AL, Watson G, et al. Shot in the dark. Surv Ophthalmol. 2023;68:821-829.
Francesca Bosello, Damiano Marastoni, Francesca Benedetta Pizzini, Chiara Zaffalon, Andrea Zuliani, Giulia Turri, Sara Mariotto, Erika Bonacci, et al. Atypical myelin oligodendrocyte glycoprotein antibody–associated optic neuritis and acute demyelinating polyneuropathy after SARS-CoV-2 infection: Case report and literature review. Journal of Neuroimmunology. 2023;375:578011. doi:10.1016/j.jneuroim.2022.578011.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. Journal of neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0. PMID:27793206; PMCID:PMC5086042.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. Journal of neurology, neurosurgery, and psychiatry. 2018;89(2):127-137. doi:10.1136/jnnp-2017-316880. PMID:29142145; PMCID:PMC5800335.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. MOG antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.