โรคประสาทตาอักเสบ เป็นโรคอักเสบของเส้นประสาทตา จากกลไกภูมิต้านตนเอง อาการหลักคือการมองเห็น ลดลงเฉียบพลันข้างเดียวและปวดเมื่อขยับลูกตาอายุที่พบบ่อยคือ 15–45 ปี พบในผู้หญิงมากกว่า อุบัติการณ์รายปีในญี่ปุ่นคือ 1.6 ต่อผู้ใหญ่ 100,000 คน
มากกว่า 90% ของโรคประสาทตาอักเสบ ชนิดไม่ทราบสาเหตุฟื้นตัวเป็นความคมชัดของภาพ 0.5 หรือดีกว่าภายใน 1 ปีหลังจากเริ่มมีอาการ และมากกว่า 70% ดีขึ้นเป็น 1.0 หรือดีกว่า (ONTT)
ทางกายวิภาคแบ่งเป็นโรคประสาทตาอักเสบ ส่วนหน้า (มีหัวประสาทตาบวม พบประมาณ 50% ในญี่ปุ่น) และโรคประสาทตาอักเสบ ส่วนหลังลูกตา
ความเสี่ยงในการเปลี่ยนเป็น MS : 25% หลังจาก 15 ปีหากไม่มีรอยโรคทำลายปลอกไมอีลิน ใน MRI สมอง, 78% หากมีรอยโรค
การรักษาทางเลือกแรกคือการให้สเตียรอยด์ แบบพัลส์ด้วย methylprednisolone 1,000 มก./วัน นาน 3 วัน การรักษาด้วยสเตียรอยด์ ชนิดรับประทานเพียงอย่างเดียวมีข้อห้ามเนื่องจากเพิ่มอัตราการกลับเป็นซ้ำ
การตรวจพบแอนติบอดีต่อ AQP4 คิดเป็นประมาณ 10% ของกรณีไม่ทราบสาเหตุและดื้อต่อสเตียรอยด์ การตรวจพบแอนติบอดีต่อ MOG ตอบสนองต่อสเตียรอยด์ แต่มักกลับเป็นซ้ำ การตรวจแอนติบอดีตามชนิดของโรคและกลยุทธ์การรักษามีความสำคัญ
โรคประสาทตาอักเสบ เป็นโรคที่เส้นประสาทตา อักเสบจากสาเหตุใดสาเหตุหนึ่ง ทำให้การทำงานของการมองเห็น ลดลง หลังจากแยกโรคเส้นประสาทตา จากการติดเชื้อและไม่ติดเชื้อ (จากพิษ พันธุกรรม การกดทับ) ออกแล้ว กรณีส่วนใหญ่เป็นชนิดไม่ทราบสาเหตุ ซึ่งสันนิษฐานว่าเกิดจากกลไกภูมิต้านตนเอง
อายุที่พบบ่อยคือ 15–45 ปี พบในผู้หญิงมากกว่า และแสดงเป็นโรคเส้นประสาทตา เฉียบพลันข้างเดียว การมองเห็น แย่ลงในช่วงสองสามวันถึง 2 สัปดาห์ จากนั้นมีแนวโน้มฟื้นตัวภายใน 5 สัปดาห์
โรคประสาทตาอักเสบ ส่วนหน้า (papillitis)โรคประสาทตาอักเสบ ส่วนหลังลูกตา
ชนิดของโรค ลักษณะ ไม่ทราบสาเหตุ (กลไกภูมิต้านตนเอง) พบบ่อยที่สุด โดยทั่วไปเป็นแบบเฟสเดียว ผลบวกต่อแอนติบอดี AQP4 (เกี่ยวข้องกับ NMOSD ) ประมาณ 10% ของกรณีไม่ทราบสาเหตุ ดื้อต่อสเตียรอยด์ อัตราส่วนเพศ 1:9 (ชาย:หญิง) ผลบวกต่อแอนติบอดี MOG (MOGAD ) เป็นทั้งสองข้าง, มี papilledema, รอยโรคยาว ตอบสนองต่อสเตียรอยด์ แต่มักกลับเป็นซ้ำ เกี่ยวข้องกับ MS ผู้ป่วย MS มากถึง 75% เคยมี ON อย่างน้อยหนึ่งครั้งในชีวิต CRION ขึ้นกับสเตียรอยด์ และกลับเป็นซ้ำ เสนอโดย Kidd และคณะในปี 20035) ติดเชื้อ ซิฟิลิส, ไวรัส ฯลฯ ต้องแยกออกก่อนใช้สเตียรอยด์
ในการตรวจครั้งแรก ให้วัดแอนติบอดี AQP4; ถ้าผลลบ ให้รักษาเป็นไม่ทราบสาเหตุ
อุบัติการณ์รายปีในญี่ปุ่นคือ 1.6 ต่อผู้ใหญ่ 100,000 คน อายุที่พบบ่อยคือ 15–45 ปี ผู้หญิงประมาณ 70% ความชุกทั่วโลกประมาณ 1–5 ต่อ 100,000 คน
ความสัมพันธ์กับ MS นั้นลึกซึ้งมาก ความน่าจะเป็นสะสมของการเปลี่ยนเป็น MS ภายใน 15 ปีหลังจากเริ่มมีภาวะเส้นประสาทตา อักเสบชนิดไม่ทราบสาเหตุคือ 50% หากไม่มีรอยโรคทำลายไมอีลินใน MRI สมองเมื่อเริ่มมีอาการครั้งแรก อัตราการเปลี่ยนจะอยู่ที่เพียง 25% แต่ถ้ามีรอยโรคอย่างน้อยหนึ่งรอย อัตราจะสูงถึง 78%
ในเด็ก อายุที่เริ่มมีอาการบ่อยที่สุดคือ 9-10 ปี และยิ่งเด็กอายุน้อยเท่าใด ก็ยิ่งพบการเกี่ยวข้องของตาทั้งสองข้างและการสูญเสียการมองเห็น อย่างรุนแรงบ่อยขึ้น โดยเฉพาะอย่างยิ่งในเด็กอายุต่ำกว่า 5 ปี อัตราการเปลี่ยนเป็น MS ในเด็กมีรายงานประมาณ 30% โดย Adachi และ 16% โดย Mizota
อุบัติการณ์รายปีของ MOGAD ประมาณ 1.6 ถึง 4.8 ต่อล้านคน 1)
Q
ภาวะเส้นประสาทตาอักเสบจะนำไปสู่โรคปลอกประสาทเสื่อมแข็งในอนาคตหรือไม่?
A
เชื่อว่าประมาณ 50% ของภาวะเส้นประสาทตา อักเสบชนิดไม่ทราบสาเหตุจะเปลี่ยนเป็น MS ภายใน 15 ปี อย่างไรก็ตาม หากไม่มีรอยโรคทำลายไมอีลินใน MRI สมองเมื่อเริ่มมีอาการครั้งแรก อัตราการเปลี่ยนจะอยู่ที่เพียง 25% หลังจากเริ่มมีอาการ การประเมินความเสี่ยงด้วย MRI สมองและการติดตามผลร่วมกับแพทย์ระบบประสาทเป็นสิ่งสำคัญ
Wikimedia Commons. File:Optic_Neuritis.png. License: CC BY-SA.
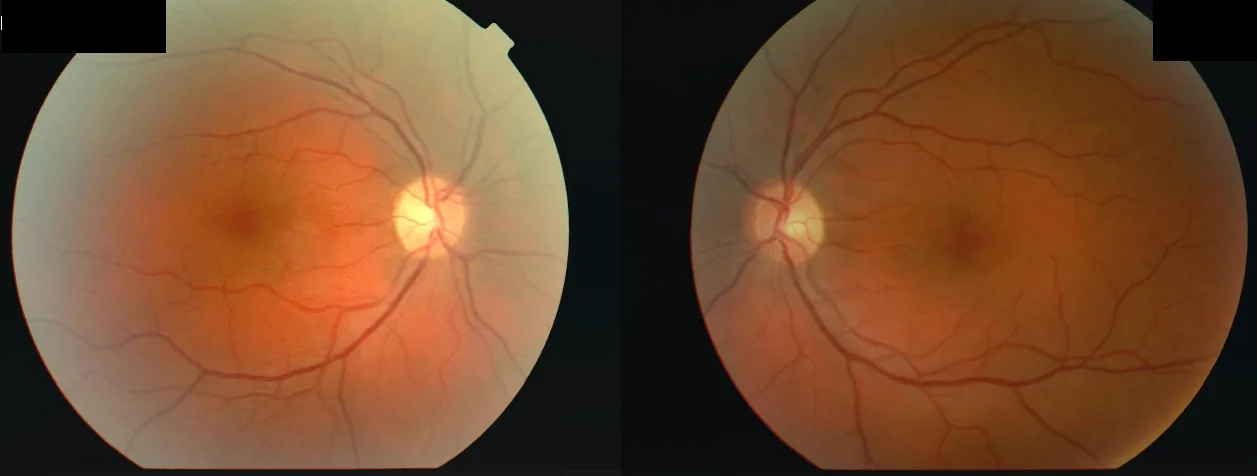
ภาพถ่ายจอตาทั้งสองข้างของผู้หญิงอายุ 34 ปีที่มี
การมองเห็น ลดลงในตาขวา (OD 2/60) ตามด้วยตาซ้าย (OS 6/24) หลังจากป่วยเป็นไข้ โดยมี
จุดบอดกลาง ตาทั้งสองข้างและความบกพร่องของ
รูม่านตา ชนิดรับสัญญาณสัมพัทธ์ (
RAPD ) ในตาขวา ซึ่งสอดคล้องกับ “
จุดบอดกลาง ตา” ที่กล่าวถึงในหัวข้อ “2. อาการหลักและผลการตรวจทางคลินิก”
มีรูปแบบการเริ่มต้นแบบเฉียบพลัน โดยมีอาการต่อไปนี้เป็นลักษณะเฉพาะ:
การมองเห็น ลดลงเฉียบพลันข้างเดียวการมองเห็น แย่ที่สุดประมาณ 1 สัปดาห์หลังจากเริ่มมีอาการปวดตา และปวดเมื่อขยับลูกตาการมองเห็น ลดลงความผิดปกติของลานสายตา : จุดบอดกลาง ตาและจุดบอดจุดบอดกลาง ตาพบได้บ่อย แต่ก็มีกรณีตาบอดครึ่งซีกแนวนอน ความผิดปกติแบบกระจายก็พบได้บ่อยเช่นกันความผิดปกติของการมองเห็นสี สัญญาณ Uhthoff : การมองเห็น ลดลงชั่วคราวหรือตามัวเมื่ออุณหภูมิร่างกายสูงขึ้น (หลังอาบน้ำหรือออกกำลังกาย) ปรากฏภายในไม่กี่นาทีและหายไปภายในหนึ่งชั่วโมงภาพแสงวาบ (phosphenes) : การรับรู้แสงแฟลชปรากฏการณ์พัลฟริช (Pulfrich phenomenon)
ความบกพร่องของรูม่านตา ชนิดรับแสงสัมพัทธ์ (RAPD ) : อาการแสดงที่สำคัญที่สุด ให้ผลบวกในกรณีที่เกิดกับตาข้างเดียวหรือไม่สมมาตร แสดงความผิดปกติแม้การทำงานบกพร่องเพียงเล็กน้อยในโรคประสาทอักเสบแก้วนำแสง ตรวจด้วยเลนส์หน้าและกล้องจุลทรรศน์ชนิดกรีดก่อนขยายรูม่านตา ระดับความบกพร่องทางการมองเห็น : ตามการศึกษา ONTT พบว่า 10% มีสายตา 1.0 หรือดีกว่า 25% อยู่ระหว่าง 0.5-1.0 29% อยู่ระหว่าง 0.1-0.5 และ 36% ต่ำกว่านั้นผลการตรวจอวัยวะภายในลูกตา : ในโรคประสาทอักเสบแก้วนำแสงส่วนหน้า พบหัวประสาทตาบวมแดงและมีการเรืองแสงมากเกินไปในการตรวจหลอดเลือดด้วยฟลูออเรสซีน ในโรคประสาทอักเสบแก้วนำแสงส่วนหลังลูกตา จะปกติในช่วงแรก ต่อมาซีดลงหลังจาก 4-6 สัปดาห์อัมพาตกล้ามเนื้อตาระหว่างนิวเคลียส (INO ) : เกิดขึ้นในกรณีที่ร่วมกับโรค MS เนื่องจากการทำลายไมอีลินของฟาสซิคูลัสตามยาวตรงกลางการเกิดปลอกหุ้มหลอดเลือดจอประสาทตา และม่านตาอักเสบ ส่วนปลาย : พบในผู้ป่วย MS 5-10%
หากมีอาการแสดงต่อไปนี้ ให้สงสัยโรคประสาทอักเสบแก้วนำแสงชนิดไม่ปกติและดำเนินการค้นหาสาเหตุอย่างละเอียด
อายุอยู่นอกช่วง 15-45 ปี (น้อยกว่า 18 หรือมากกว่า 50)
เกิดพร้อมกันทั้งสองข้าง
อาการดำเนินต่อเนื่องหลังจากเริ่มมีอาการ 2 สัปดาห์
การดำเนินโรคที่ต้องพึ่งสเตียรอยด์
มีอาการทางระบบร่วมด้วย
ลักษณะ MOG-ON AQP4-ON MS -ONCRION อัตราส่วนเพศ (ชาย:หญิง) 1:1 1:7-9 1:3 1:1.7-2.3 การเป็นสองตา 31-84% 13-82% พบน้อยมาก มี ปุ่มประสาทตาบวม 45-92% 7-52% 11-14% เล็กน้อย ปวดตา 73-92% 28-50% 10-46% มี การมองเห็น แย่ที่สุดปานกลางถึงรุนแรง ปานกลางถึงรุนแรง เล็กน้อยถึงปานกลาง หลากหลาย การฟื้นตัว ดี อาจแย่ ดี พึ่งพาสเตียรอยด์
ที่มา: Jeyakumar N, et al. Eye. 20241)
ในโรคประสาทอักเสบแก้วนำแสงในเด็ก มักแสดงเป็น papillitis ทั่วไปที่มีรอยแดงและบวมของจานประสาทตา แทนที่จะเป็น retrobulbar neuritis ที่พบในผู้ใหญ่ การอักเสบไม่ได้จำกัดอยู่ที่การบวมของ papilla แต่กระจายไปตามความยาวทั้งหมดของเส้นประสาทตา การเป็นสองข้างพบได้ 50-75% และชนิด anterior optic neuritis คิดเป็น 50-75%
Q
อาการของ Uhthoff sign คืออะไร?
A
Uhthoff sign เป็นปรากฏการณ์ที่เกิดการสูญเสียการมองเห็น ชั่วคราวเมื่ออุณหภูมิร่างกายสูงขึ้น ผู้ป่วยสังเกตเห็นการมองเห็น ลดลงหรือตาพร่า หลังจากอาบน้ำอุ่นหรือออกกำลังกาย อาการจะปรากฏภายในไม่กี่นาทีและหายไปภายในหนึ่งชั่วโมง เป็นที่รู้จักในโรคประสาทอักเสบแก้วนำแสงที่เกี่ยวข้องกับ MS แต่ก็มีรายงานในโรคเส้นประสาทตา อื่นๆ และไม่จำเพาะต่อ MS
Q
โรคประสาทอักเสบแก้วนำแสงที่ตรวจพบแอนติบอดีต่อ AQP4 แตกต่างจากโรคประสาทอักเสบแก้วนำแสงทั่วไปอย่างไร?
A
โรคประสาทอักเสบแก้วนำแสงที่ตรวจพบแอนติบอดีต่อ AQP4 เป็นชนิดดื้อยาซึ่งคิดเป็นประมาณ 10% ของ idiopathic optic neuritis เมื่อเทียบกับโรคประสาทอักเสบแก้วนำแสงทั่วไป การมองเห็น เมื่อเริ่มเป็นจะแย่กว่า การตอบสนองต่อการรักษาด้วยสเตียรอยด์ ไม่ดี และการกลับเป็นซ้ำบ่อยกว่า นอกจากนี้ยังแตกต่างในแนวโน้มที่จะกลายเป็นสองข้าง การวินิจฉัยจำเป็นต้องตรวจวัดแอนติบอดีต่อ AQP4 ในซีรัม
ในกรณีที่ไม่ทราบสาเหตุซึ่งสงสัยว่ามีกลไก autoimmune เซลล์ที่เกี่ยวข้องกับการอักเสบ เช่น microglia จะแทรกซึมเข้าไปในเส้นประสาทตา และทำให้เกิดการอักเสบ การอักเสบซ้ำๆ ทำให้เกิดความเสียหายสะสมต่อเส้นใยประสาท นำไปสู่การฝ่อของเส้นประสาทตา
มีสมมติฐานว่าการติดเชื้อไวรัสอาจเป็นตัวกระตุ้นปฏิกิริยา autoimmune และตรวจพบแอนติบอดีต่อไวรัสหัด อีสุกอีใส ไข้หวัดใหญ่ ฯลฯ ในน้ำไขสันหลังของผู้ป่วยบางราย ในเด็ก เป็นที่ทราบกันว่ามีความสัมพันธ์กับ ADEM (acute disseminated encephalomyelitis) ซึ่งมีไข้และปวดศีรษะร่วมด้วย
เพศ : พบในผู้หญิงมากกว่า (อัตราส่วนชาย:หญิงประมาณ 1:2 ถึง 1:3)อายุ : มักเกิดในช่วงอายุ 20–45 ปีอาการนำ : อาจมีอาการคล้ายไข้หวัดใหญ่มาก่อนผู้ป่วย MS : มากถึง 75% จะมี ON อย่างน้อยหนึ่งครั้งตลอดชีวิต ในการชันสูตรพลิกศพ พบรอยโรคที่เส้นประสาทตา ได้ถึง 90%
มีรายงานว่า MOGAD เกิดหลังการฉีดวัคซีนหรือการติดเชื้อ และกลไกเช่น bystander activation ถูกสันนิษฐานว่าทำให้เกิดการสูญเสียความทนทานต่อภูมิคุ้มกัน 1)
มีรายงานการเกิด ON หลังการติดเชื้อ COVID-19 หรือการฉีดวัคซีน ค่ามัธยฐานของระยะเวลาจากการฉีดวัคซีนถึงการเกิด ON คือ 18 วัน และ 14 ใน 55 รายมีผลบวกต่อ MOG-IgG 11)
โรคประสาทตาอักเสบ เป็นการวินิจฉัยทางคลินิก วินิจฉัยจากการรวมกันของภาวะการมองเห็น ลดลงเฉียบพลันข้างเดียว ปวดเมื่อขยับตา RAPD บวก และความบกพร่องของลานสายตา
การตรวจวัดสายตา การตรวจรีเฟล็กซ์รูม่านตา (swinging flashlight test) : จำเป็นสำหรับการตรวจหา RAPD การตรวจลานสายตา จุดบอดกลาง หรือความบกพร่องของชั้นใยประสาทโดยใช้เครื่องวัดลานสายตาอัตโนมัติการตรวจการมองเห็น สีและความไวต่อคอนทราสต์ ค่าการกะพริบกลาง (CFF ) : พบว่าลดลงการตรวจอวัยวะภายในลูกตา : ตรวจสอบว่ามี papilledema หรือไม่
MRI วงโคจร : ภาพตัดแนว冠状 STIR ระงับสัญญาณไขมัน, T1 ที่มีการฉีดสารทึบรังสี ตรวจสอบการขยายใหญ่ของเส้นประสาทตา , สัญญาณสูง, และการเพิ่มความเข้มของสารทึบรังสีMRI ศีรษะ FLAIR : ประเมินว่ามีรอยโรคทำลายไมอีลินในสมองหรือไม่ จำเป็นสำหรับการประเมินโรคร่วม MS และความเสี่ยงในการเกิด MS ในอนาคต
ผล MRI ใน ON แบบปกติและแบบผิดปกติแตกต่างกันในประเด็นต่อไปนี้
ลักษณะ ON แบบปกติ (สัมพันธ์กับ MS ) ON แบบผิดปกติ รูปแบบการเพิ่มความเข้มของสารทึบรังสี การเพิ่มความเข้มแบบสั้นเป็นช่วง การเพิ่มความเข้มแบบยาวและกว้าง (≥1/2) ตำแหน่งรอยโรค ด้านหน้า (เส้นประสาทตา ส่วน retrobulbar) ขยายไปด้านหลัง (optic chiasm และ optic tract) ปลอกประสาทตา ไม่มีการเพิ่มความเข้มของสารทึบรังสี การเพิ่มความเข้มของสารทึบรังสีของปลอกประสาทตาและไขมันในเบ้าตา
การเปรียบเทียบลักษณะทางภาพของ NMOSD -ON, MOG-ON และ MS -ON:
ลักษณะ NMOSD -ONMOG-ON MS -ONรูปแบบการเกิดโรค ทั้งสองข้าง ทั้งสองข้าง ข้างเดียว ตำแหน่งที่พบบ่อย ในกะโหลกศีรษะ / ออปติกไคแอสมาอ หลังลูกตา (ส่วนหน้า) หลังลูกตา / ในคลองประสาทตา ความยาวของรอยโรค ยาว ยาว ส่วนสั้น การเพิ่มความเข้มของปลอกประสาทตา พบได้น้อย บวกใน 50% ไม่มีรายงาน
ที่มา: Jeyakumar 20241) ; Cacciaguerra & Flanagan4)
วัดความหนาของชั้นใยประสาทจอประสาทตา บริเวณรอบหัวประสาทตา (pRNFL ) ในระยะเฉียบพลัน pRNFL หนาขึ้น (ค่ามัธยฐาน MOG-ON 164 μm เทียบกับ MS -ON 103 μm) ในระยะเรื้อรัง pRNFL บางลง และจำนวนครั้งที่กลับเป็นซ้ำสัมพันธ์กับการลดลงของ pRNFL เมื่อ pRNFL ต่ำกว่าเกณฑ์ 50 μm ค่า mean deviation ของลานสายตาแย่ลงอย่างมีนัยสำคัญ1)
พบการยืดออกของระยะแฝง P100 ซึ่งสะท้อนถึงการทำลายปลอกไมอีลิน
ในกรณีที่มีแนวทางไม่ปกติ ให้วัดสิ่งต่อไปนี้:
แอนติบอดี AQP4 : ครอบคลุมโดยประกันตั้งแต่ปี 2013 การประเมินตั้งแต่เนิ่นๆ มีความสำคัญในกรณีที่ดื้อต่อสเตียรอยด์ หรือมีอาการกำเริบซ้ำแอนติบอดี MOG (MOG-IgG )
ตามเกณฑ์การวินิจฉัย MOGAD ระหว่างประเทศปี 2023 การวินิจฉัยที่แน่นอนคือ CBA บวก + รูปแบบทางคลินิกหลัก + การแยกโรคอื่นออก ในกรณีที่ไทเทอร์ต่ำ จำเป็นต้องมีลักษณะทางคลินิก/MRI ที่สนับสนุนอย่างน้อยหนึ่งอย่าง2)
การตรวจสอบเกณฑ์ดังกล่าว: ความไว 96.5%, ความจำเพาะ 98.9%, ความแม่นยำ 98.5%3)
ประเมินความเป็นไปได้ที่โรคประสาทอักเสบแก้วนำแสงจะเป็นอาการเริ่มแรกของ MS การวินิจฉัยทำได้โดยการพิสูจน์การกระจายตามเวลาและสถานที่ของรอยโรคทำลายปลอกไมอีลิน อักเสบในระบบประสาทส่วนกลาง และแยกโรคอื่นออก สามารถพิสูจน์การกระจายตามสถานที่ได้หากมีรอยโรคสัญญาณสูง T2 ในสองบริเวณขึ้นไปจากสี่บริเวณ: รอบโพรงสมอง, ใต้คอร์เทกซ์, ใต้เทนโทเรียม และไขสันหลัง
การจำแนก โรคที่ต้องแยกหลัก จุดที่แตกต่าง ทำลายปลอกไมอีลิน โรคประสาทอักเสบแก้วนำแสงที่เกี่ยวข้องกับ MS , NMO การตรวจแอนติบอดีและ MRI ภูมิคุ้มกันเป็นสื่อกลาง CRION, ซาร์คอยโดซิส , SLE การพึ่งพาสเตียรอยด์ , อาการทางระบบ ติดเชื้อ ซิฟิลิส เริม วัณโรค ต้องแยกออกก่อนใช้สเตียรอยด์ พันธุกรรม โรคเส้นประสาทตาถ่ายทอดทางพันธุกรรมชนิด Leber ชายหนุ่ม ตาทั้งสองข้าง ไม่เจ็บปวด → ตรวจยีน จากยา เอแทมบูทอล ลิเนโซลิด ไม่มีการเพิ่มความเข้มของสารทึบรังสีใน MRI หลอดเลือด โรคเส้นประสาทตาขาดเลือดส่วนหน้าชนิดไม่สัมพันธ์กับหลอดเลือดแดงอักเสบ ผู้สูงอายุ ปกติไม่เจ็บปวด จานประสาทตา บวมซีดบางส่วน เด็ก ADEM ร่วมกับไข้ ปวดหัว สติสัมปชัญญะผิดปกติ
มีรายงานผู้ป่วยที่ได้รับการวินิจฉัยว่าเป็นโรคประสาทตาอักเสบ ชนิด retrobulbar แต่แท้จริงแล้วเป็นโรคเส้นประสาทตาถูกกดทับ จากมะเร็งต่อมน้ำเหลืองในเบ้าตา ดังนั้นหากมีลักษณะผิดปกติ ไม่ควรให้สเตียรอยด์ อย่างง่ายดาย และควรตรวจสอบอย่างละเอียด 9)
โดยทั่วไป การตรวจ FA (การถ่ายภาพหลอดเลือดด้วยฟลูออเรสซีน ), CFF และลานสายตาแบบพลวัตที่ทำในผู้ใหญ่ไม่เหมาะกับเด็ก การวินิจฉัยอาศัยผลการตรวจอวัยวะรับภาพและ MRI ศีรษะ
Q
จุดที่แยกโรคประสาทตาอักเสบแบบทั่วไปและแบบผิดปกติใน MRI คืออะไร?
A
ใน ON แบบทั่วไป จะพบการเพิ่มความเข้มของสัญญาณเฉพาะในส่วนสั้นของเส้นประสาทตา ในขณะที่ ON แบบผิดปกติจะพบการเพิ่มความเข้มของสัญญาณยาวของเส้นประสาทตา (มากกว่าครึ่งหนึ่งของความยาว) การขยายไปทางด้านหลัง (ออปติกไคแอสมาออปติกแทรกต์) และการเพิ่มความเข้มของสัญญาณปลอกหุ้มเส้นประสาทตา ผล MRI เหล่านี้เชื่อมโยงโดยตรงกับการวินิจฉัยแยกโรคที่เป็นสาเหตุ
การพยากรณ์การทำงานของการมองเห็น ในโรคประสาทตาอักเสบ ชนิดไม่ทราบสาเหตุนั้นดี ผู้ป่วยโรคประสาทตาอักเสบ มากกว่า 90% มีการมองเห็น ดีขึ้นด้วยการสังเกตหรือการให้สเตียรอยด์ ทั้งระบบ ตามผลระยะยาวของ ONTT ผู้ป่วย 93% มีค่าสายตา 0.5 ขึ้นไปหลังจาก 1 ปีนับจากเริ่มป่วย และมากกว่า 70% มีค่าสายตา 1.0 ขึ้นไป
ในโรคประสาทตาอักเสบ ชนิดไม่ทราบสาเหตุที่มีค่าสายตาแก้ไขค่อนข้างดี สามารถสังเกตอาการโดยรับประทานเมโคบาลามิน 1500 ไมโครกรัม/วัน (นอกเหนือสิทธิ์ประกัน)
ON แบบทั่วไป
ทางเลือกแรก : การรักษาด้วยสเตียรอยด์แบบพัลส์ (methylprednisolone 1000 มก./วัน ฉีดเข้าหลอดเลือดดำ 3 วัน) นอกเหนือสิทธิ์ประกัน
หลังพัลส์ : เริ่มรับประทาน prednisolone 0.5 มก./กก./วัน แล้วค่อยๆ ลดลง 5-10 มก. ทุก 3-4 วัน
หากไม่ตอบสนองต่อพัลส์ครั้งแรก : ให้พัลส์ครั้งที่สองหลังจาก 4-5 วัน
ผล : ทำให้ระยะเวลาฟื้นตัวสั้นลง แต่ไม่มีความแตกต่างอย่างมีนัยสำคัญในค่าสายตาสุดท้ายหลังจาก 1 ปี
ข้อบ่งชี้ที่ต้องรักษา : ตาทั้งสองข้างมีอาการ, ความบกพร่องทางการมองเห็น อย่างรุนแรง, ตาข้างเดียวที่ใช้งานได้, กรณีที่กลับเป็นซ้ำ, มีรอยโรคทำลายไมอีลินใน MRI, ผู้ป่วยต้องการให้อาการดีขึ้นเร็ว
โรคเส้นประสาทตาอักเสบที่ตรวจพบแอนติบอดี AQP4
ระยะเฉียบพลัน : การรักษาด้วยสเตียรอยด์ แบบชีพจร (ทางเลือกแรก)
เมื่อไม่ได้ผล : ให้ชีพจรซ้ำอีกครั้งหลังจาก 3-4 วัน → หากยังไม่ได้ผล → พิจารณาการแลกเปลี่ยนพลาสมา
การแลกเปลี่ยนพลาสมา การแลกเปลี่ยนพลาสมา อย่างง่าย > การแลกเปลี่ยนพลาสมา แบบเมมเบรนคู่ > การดูดซับภูมิคุ้มกัน (ตามลำดับประสิทธิผล) หนึ่งคอร์ส 5-6 ครั้ง
การรักษาระยะยาว : เพรดนิโซโลน 5-10 มก./วัน + อะซาไธโอพรีน 50-100 มก./วัน
ข้อควรระวัง : แตกต่างจากโรคเส้นประสาทตา อักเสบที่ไม่ทราบสาเหตุ การป้องกันการกลับเป็นซ้ำด้วยสเตียรอยด์ ชนิดรับประทานมีความสำคัญ
โรคเส้นประสาทตาอักเสบที่ตรวจพบแอนติบอดี MOG (MOGAD)
ระยะเฉียบพลัน : การรักษาด้วยสเตียรอยด์ แบบชีพจรมีประสิทธิภาพสูง หายสมบูรณ์ 50% หายบางส่วน 44% 13)
แนวโน้มการกลับเป็นซ้ำ : การพึ่งพาสเตียรอยด์ สูง 70% กลับเป็นซ้ำระหว่างการลดขนาดเพรดนิโซโลนชนิดรับประทาน (โดยเฉพาะน้อยกว่า 10 มก./วัน หรือภายใน 2 เดือนหลังจากหยุด) 14)
การรักษาระยะยาว : IVIg (≥1 กรัม/กก./4 สัปดาห์ช่วยลดการกลับเป็นซ้ำได้อย่างมีนัยสำคัญ) 15) ริทูซิแมบ อาจมีประสิทธิภาพน้อยกว่าใน AQP4-ON
เวลาเริ่มการรักษา : โดยปกติหลังการกำเริบครั้งที่สอง (เนื่องจากมากกว่า 50% เป็นแบบครั้งเดียว)
CRION (โรคเส้นประสาทตาอักเสบเรื้อรังแบบกลับเป็นซ้ำ)
ระยะเฉียบพลัน : เมทิลเพรดนิโซโลนทางหลอดเลือดดำ 1 มก./กก. × 3-5 วัน
ระยะกลาง : เพรดนิโซนชนิดรับประทาน 1 มก./กก. จากนั้นลดขนาดลงทีละน้อยจนถึงขนาดที่มีประสิทธิภาพต่ำสุด
การรักษาระยะยาว : ยาที่เป็นตัวเลือก ได้แก่ azathioprine, methotrexate, cyclophosphamide, mycophenolate mofetil และ IVI G ก็เป็นทางเลือกหนึ่ง
ข้อควรระวัง : เนื่องจากอาจเกิดการกลับเป็นซ้ำเมื่อหยุดการรักษา จึงจำเป็นต้องรักษาต่อเนื่องในระยะยาว
การรักษาด้วย prednisolone ชนิดรับประทานขนาดมาตรฐาน (1 มก./กก./วัน) เพียงอย่างเดียวไม่แนะนำ เนื่องจากในการศึกษา ONTT พบว่ามีอัตราการกลับเป็นซ้ำสูงกว่ายาหลอกหรือสเตียรอยด์ ทางหลอดเลือดดำ
ให้ methylprednisolone ขนาด 30 มก./กก./วัน นาน 3 วัน การพยากรณ์โรคทางสายตาโดยทั่วไปดี
ให้การรักษาด้วยสเตียรอยด์ อย่างเข้มข้น หากตอบสนองต่อสเตียรอยด์ แบบ pulse ไม่ดี อาจพิจารณาให้อิมมูโนโกลบูลินทางหลอดเลือดดำขนาดสูงหรือการแลกเปลี่ยนพลาสมา
ในกรณีที่มี MS ร่วมด้วย หลังจากสายตาดีขึ้น ให้พิจารณาการรักษาเพื่อป้องกันการกลับเป็นซ้ำ โดยประสานงานกับอายุรแพทย์ระบบประสาท พิจารณายาปรับเปลี่ยนโรค (เช่น interferon beta, glatiramer acetate, fingolimod, natalizumab เป็นต้น)
Q
การรักษาด้วยสเตียรอยด์ส่งผลต่อการฟื้นฟูการมองเห็นขั้นสุดท้ายหรือไม่?
A
ผลลัพธ์ของ ONTT แสดงให้เห็นว่าการรักษาด้วยสเตียรอยด์แบบพัลส์ ช่วยเพิ่มความเร็วในการฟื้นตัว แต่ไม่มีความแตกต่างอย่างมีนัยสำคัญในการมองเห็น ขั้นสุดท้ายหลังจากหนึ่งปีนับจากเริ่มมีอาการ ในโรคประสาทอักเสบแก้วนำแสงชนิดไม่ทราบสาเหตุ 93% ฟื้นตัวเป็นการมองเห็น 0.5 หรือดีกว่าโดยไม่ต้องรักษา อย่างไรก็ตาม กรณีที่ตรวจพบแอนติบอดีต่อ AQP4 เป็นบวกจะดื้อต่อสเตียรอยด์ จึงจำเป็นต้องได้รับการรักษาเชิงรุกตั้งแต่เนิ่นๆ รวมถึงการแลกเปลี่ยนพลาสมา
Q
ต้องรักษาด้วยยากดภูมิคุ้มกันต่อไปนานเท่าใด?
A
เนื่องจาก CRION เป็นโรคที่กลับเป็นซ้ำเมื่อหยุดการรักษา จึงมักจำเป็นต้องรักษาด้วยยากดภูมิคุ้มกันในระยะยาวแม้หลังจากอาการทุเลาแล้ว กำหนดขนาดยาสเตียรอยด์ ที่มีประสิทธิภาพต่ำที่สุด จากนั้นเพิ่มยากดภูมิคุ้มกันที่ไม่ใช่สเตียรอยด์ และออกแบบสูตรการรักษาเป็นรายบุคคล
ในกรณีที่ไม่ทราบสาเหตุซึ่งสงสัยว่ามีกลไกภูมิต้านตนเอง เซลล์ที่เกี่ยวข้องกับการอักเสบ เช่น ไมโครเกลีย จะแทรกซึมเข้าไปในเส้นประสาทตา และทำให้เกิดการอักเสบ
การทำลายปลอกไมอีลินโดยอาศัยภูมิคุ้มกัน : ปลอกไมอีลินของเส้นประสาทตา ถูกโจมตีโดยกลไกภูมิต้านตนเอง การนำกระแสประสาทแบบกระโดดไม่สามารถเกิดขึ้นได้ ทำให้เกิดความผิดปกติในการนำกระแสประสาทตามแนวแอกซอนการเสื่อมของแอกซอน : หลังจากการทำลายปลอกไมอีลิน แอกซอนของเซลล์ปมประสาทจอประสาทตา เริ่มเสื่อมการกินของเสียโดยแมคโครฟาจ : แมคโครฟาจกำจัดเศษไมอีลินที่เหลืออยู่ไกลโอซิส : แอสโตรไซต์เพิ่มจำนวนและสร้างแผลเป็นเกลีย คำว่า “เส้นโลหิตตีบ” ในโรคปลอกประสาทเสื่อมแข็งมีที่มาจากสิ่งนี้
เมื่อเกิดการอักเสบของเส้นประสาทตา ซ้ำๆ ความเสียหายสะสมต่อเส้นใยประสาทจะนำไปสู่การฝ่อของเส้นประสาทตา
แอนติบอดีต่อ AQP4 จับกับคอมพลีเมนต์ และโจมตีแอสโตรไซต์ (เซลล์เกลีย) ภายในเส้นประสาทตา ทำให้เกิดโรค แอสโตรไซต์ในเส้นประสาทตา และออปติกไคแอสมาสแสดง AQP4 จำนวนมาก ทำให้เป็นเป้าหมายที่ง่าย
MOG-IgG (ส่วนใหญ่เป็นคลาสย่อย IgG1) จับกับ MOG บนผิวของปลอกไมอีลินในระบบประสาทส่วนกลาง การกระตุ้นคอมพลีเมนต์ และฟาโกไซโทซิสที่ขึ้นกับแอนติบอดีมีส่วนร่วมในการทำลายไมอีลิน อย่างไรก็ตาม การกระตุ้นคอมพลีเมนต์ อ่อนแอกว่าเมื่อเทียบกับ AQP4-IgG ใน MOGAD เซลล์ที CD4-positive และมาโครฟาจเป็นหลัก ในขณะที่ใน MS เซลล์ที CD8-positive เป็นหลัก 1) 4)
แม้ว่า MOG จะไม่แสดงออกในจอประสาทตา แต่โรคประสาทตาอักเสบ ที่เกี่ยวข้องกับ MOG ทำให้เกิดความเสียหายต่อเซลล์ปมประสาทจอประสาทตา มีการเสนอว่าพิษของกลูตาเมตและความเปราะบางของอุปสรรคเลือด-สมองที่หัวประสาทตาเป็นกลไก 1)
IL-6 เพิ่มการซึมผ่านของอุปสรรคเลือด-สมองและส่งเสริมการเปลี่ยนแปลงของพลาสมาบลาสต์ ซึ่งเป็นกลไกที่กำลังได้รับความสนใจเช่นกัน
CRION ถูกเสนอครั้งแรกในฐานะกลุ่มอาการที่ครอบคลุมโรคประสาทตาอักเสบ ที่ตอบสนองต่อสเตียรอยด์ และมีแนวโน้มกำเริบ 5) การตรวจแอนติบอดีในกลุ่มผู้ป่วย CRION ภายหลังพบว่าสูงถึง 22% เป็นบวกต่อ AQP4-IgG และสูงถึง 25% เป็นบวกต่อ MOG-IgG 6) 7) 8) ดังนั้น CRION จึงถือเป็นการวินิจฉัยแบบกลุ่มอาการที่รวมกลุ่มสาเหตุที่แตกต่างกัน รวมถึงโรคประสาทตาอักเสบที่เกี่ยวข้องกับแอนติบอดี MOG และโรคประสาทตาอักเสบ ที่เกี่ยวข้องกับแอนติบอดี AQP4
มีการเสนอสมมติฐานว่าการติดเชื้อไวรัสอาจเป็นตัวกระตุ้นให้เกิดการตอบสนองทางภูมิคุ้มกันตนเอง มีรายงานการตรวจพบ DNA ของไวรัส varicella-zoster และไวรัสเริมในน้ำไขสันหลังของผู้ป่วยโรคประสาทตาอักเสบ บางราย ใน MOGAD การฉีดวัคซีนหรือการติดเชื้อถูกสันนิษฐานว่าทำให้เกิดการทำลายความทนทานต่อภูมิคุ้มกัน (การกระตุ้นแบบ bystander หรือการเลียนแบบโมเลกุล) 1)
ยาชีววัตถุ ที่มีหลักฐานระดับ 1 สำหรับ NMOSD ที่เป็นบวกต่อ AQP4 ได้เกิดขึ้นแล้ว 4)
Eculizumab : ตัวยับยั้ง C5 ระยะที่ 3 แสดงการลดความเสี่ยงของการกำเริบของโรค 94%Ravulizumab : ตัวยับยั้ง C5 (รุ่นปรับปรุงของ eculizumab) ระยะที่ 3 แสดงการลดความเสี่ยงของการกำเริบของโรค 98.6%Inebilizumab : ยากำจัดเซลล์บีที่กำหนดเป้าหมาย CD19 ลดความเสี่ยงการกลับเป็นซ้ำ 77% ใน NMOSD ที่มี AQP4 บวกSatralizumab : ตัวยับยั้งตัวรับ IL-6 ลดความเสี่ยงการกลับเป็นซ้ำ 74-79% ใน NMOSD ที่มี AQP4 บวก สามารถฉีดใต้ผิวหนังที่บ้านได้
ยาเหล่านี้ทั้งหมดมีหลักฐานที่ชัดเจนสำหรับ NMOSD ที่มี AQP4 บวก แต่ยังไม่มีการกำหนดข้อบ่งชี้สำหรับ MOGAD
การทดลองทางคลินิกระยะที่ 3 (RCT) กำลังดำเนินการสำหรับ satralizumab (NCT 05271409) และ rozanolixizumab (NCT 05063162) การใช้นอกเหนือข้อบ่งชี้ของ tocilizumab (แอนติบอดีต่อตัวรับ IL-6) รายงานผลการป้องกันการกลับเป็นซ้ำนานถึง 29 เดือน 1)
มีรายงานผู้ป่วยโรคประสาทอักเสบแก้วนำแสงแบบทำลายปลอกไมอีลินหลังการติดเชื้อ COVID-19 Jossy และคณะ (2022) รายงานผู้ป่วยที่เกิดโรคประสาทอักเสบแก้วนำแสงในช่วงพักฟื้น และทุกรายฟื้นการมองเห็น ด้วยการรักษาด้วยสเตียรอยด์ แบบชีพจร 10)
รายงานผู้ป่วยโรคประสาทอักเสบแก้วนำแสงหลังการฉีดวัคซีน COVID-19 สะสมมากขึ้น โดยมีค่ามัธยฐานของการเริ่มต้น 18 วันหลังฉีดวัคซีน ใน 55 ราย มี 14 รายที่ตรวจพบ MOG-IgG บวก และไม่มีรายใดที่ตรวจพบ AQP4-IgG บวก 11) นอกจากนี้ยังมีรายงานผู้ป่วยโรคประสาทอักเสบแก้วนำแสงที่เกี่ยวข้องกับ MOG หลังการติดเชื้อ SARS-CoV-2 12)
ด้วยการแพร่หลายของการตรวจแอนติบอดี MOG -IgG และ AQP4-IgG ผู้ป่วยบางรายที่เคยได้รับการวินิจฉัยว่าเป็น CRION กำลังถูกจัดประเภทใหม่เป็น MOGAD หรือ NMOSD คาดว่าการนิยามแนวคิดโรคใหม่ตามโปรไฟล์แอนติบอดีจะก้าวหน้าต่อไป
Jeyakumar N, Lerch M, Dale RC, Ramanathan S. MOG antibody-associated optic neuritis. Eye (London, England). 2024;38(12):2289-2301. doi:10.1038/s41433-024-03108-y. PMID:38783085; PMCI D:PMC11306565.
Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: International MOGAD Panel proposed criteria. The Lancet. Neurology. 2023;22(3):268-282. doi:10.1016/S1474-4422(22)00431-8. PMID:36706773.
Varley JA, Champsas D, Prossor T, et al. Validation of the 2023 International Diagnostic Criteria for MOGAD in a Selected Cohort of Adults and Children. Neurology. 2024.
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment: A Tale of Two Central Nervous System Autoimmune Inflammatory Disorders. Neurol Clin. 2024;42(1):77-114. doi:10.1016/j.ncl.2023.06.009. PMID:37980124; PMCI D:PMC10658081.
Kidd D, Burton B, Plant GT, Graham EM. Chronic relapsing inflammatory optic neuropathy (CRION). Brain. 2003;126:276-84. doi:10.1016/s0002-9394(03)00281-2.
Petzold A, Woodhall M, Khaleeli Z, Tobin WO, Pittock SJ, Weinshenker BG, et al. Aquaporin-4 and myelin oligodendrocyte glycoprotein antibodies in immune-mediated optic neuritis at long-term follow-up. Journal of neurology, neurosurgery, and psychiatry. 2019;90(9):1021-1026. doi:10.1136/jnnp-2019-320493. PMID:31118222.
Lee HJ, Kim B, Waters P, Woodhall M, Irani S, Ahn S, et al. Chronic relapsing inflammatory optic neuropathy (CRION): a manifestation of myelin oligodendrocyte glycoprotein antibodies. Journal of neuroinflammation. 2018;15(1):302. doi:10.1186/s12974-018-1335-x. PMID:30382857; PMCI D:PMC6208174.
Petzold A, Pittock S, Lennon V, Maggiore C, Weinshenker BG, Plant GT. Neuromyelitis optica-IgG (aquaporin-4) autoantibodies in immune mediated optic neuritis. Journal of neurology, neurosurgery, and psychiatry. 2010;81(1):109-11. doi:10.1136/jnnp.2008.146894. PMID:20019228.
McElhinney K, Rhatigan M, Tsvetanova Z, O’Keane C, Logan P. Beware the Retrobulbar Optic Neuritis Diagnosis. Case reports in ophthalmology. 2022;13(2):453-458. doi:10.1159/000524685. PMID:35950025; PMCI D:PMC9247490.
Jossy A, Jacob N, Sarkar S, Gokhale T, Kaliaperumal S, Deb AK. COVID-19-associated optic neuritis - A case series and review of literature. Indian journal of ophthalmology. 2022;70(1):310-316. doi:10.4103/ijo.IJO_2235_21. PMID:34937266; PMCI D:PMC8917537.
Bhatti MT, Gilbert AL, Watson G, et al. Shot in the dark. Surv Ophthalmol. 2023;68:821-829.
Francesca Bosello, Damiano Marastoni, Francesca Benedetta Pizzini, Chiara Zaffalon, Andrea Zuliani, Giulia Turri, Sara Mariotto, Erika Bonacci, et al. Atypical myelin oligodendrocyte glycoprotein antibody–associated optic neuritis and acute demyelinating polyneuropathy after SARS-CoV-2 infection: Case report and literature review. Journal of Neuroimmunology. 2023;375:578011. doi:10.1016/j.jneuroim.2022.578011.
Jarius S, Ruprecht K, Kleiter I, Borisow N, Asgari N, Pitarokoili K, Pache F, Stich O, et al. MOG-IgG in NMO and related disorders: a multicenter study of 50 patients. Part 2: Epidemiology, clinical presentation, radiological and laboratory features, treatment responses, and long-term outcome. Journal of neuroinflammation. 2016;13(1):280. doi:10.1186/s12974-016-0718-0. PMID:27793206; PMCI D:PMC5086042.
Ramanathan S, Mohammad S, Tantsis E, Nguyen TK, Merheb V, Fung VSC, et al. Clinical course, therapeutic responses and outcomes in relapsing MOG antibody-associated demyelination. Journal of neurology, neurosurgery, and psychiatry. 2018;89(2):127-137. doi:10.1136/jnnp-2017-316880. PMID:29142145; PMCI D:PMC5800335.
Chen JJ, Flanagan EP, Jitprapaikulsan J, et al. MOG antibody-positive optic neuritis: clinical characteristics, radiologic clues, and outcome. Am J Ophthalmol. 2018;195:8-15.