โรคกลุ่มอาการอักเสบของเส้นประสาทตา และไขสันหลัง (NMOSD ) เป็นโรคภูมิต้านตนเองของระบบประสาทส่วนกลาง ซึ่งส่วนใหญ่มีแอนติบอดีต่ออะควาพอริน-4 (AQP4) เป็นสื่อกลาง
มีลักษณะเด่นคือเส้นประสาทตา อักเสบและไขสันหลังอักเสบ พบได้บ่อยในเพศหญิง (อัตราส่วนเพศประมาณ 1:9) วัยกลางคน และเชื้อสายเอเชีย
เส้นประสาทตา อักเสบใน NMOSD รุนแรงกว่า เป็นสองข้าง และกลับเป็นซ้ำบ่อยกว่าเมื่อเทียบกับ MS และทำให้สูญเสียการมองเห็น ถาวรอย่างน้อย 20/200 ในตาข้างเดียวใน 60-69% ของกรณีไขสันหลังอักเสบมีลักษณะเฉพาะคือรอยโรคตามยาวกว้าง (LETM) ที่ยาวตั้งแต่สามปล้องกระดูกสันหลังขึ้นไป และทำให้เกิดกลุ่มอาการไขสันหลังสมบูรณ์ (ความผิดปกติด้านการเคลื่อนไหว ความรู้สึก และระบบประสาทอัตโนมัติ)
การวินิจฉัยขึ้นอยู่กับการตรวจวัด AQP4-IgG (แนะนำให้ใช้การทดสอบโดยใช้เซลล์เป็นฐาน) และ MRI เป็นหลัก
ในระยะเฉียบพลัน ใช้การให้สเตียรอยด์ ขนาดสูงทางหลอดเลือดดำ หากตอบสนองไม่เพียงพอ การแลกเปลี่ยนพลาสมา คือทางเลือกถัดไป
เพื่อป้องกันการกลับเป็นซ้ำ ใช้ยายับยั้งคอมพลีเมนต์ (อีคูลิซูแมบ) การบำบัดด้วยการกำจัดเซลล์บี (ริทูซิแมบ ) และยายับยั้งตัวรับ IL-6 (ซาทราลิซูแมบ) และแนะนำให้เริ่มตั้งแต่เนิ่นๆ หลังการเกิดอาการครั้งแรก
โรคกลุ่มอาการอักเสบของเส้นประสาทตา และไขสันหลัง (NMOSD ) เป็นโรคอักเสบที่เกิดจากแอนติบอดีและภูมิต้านตนเอง ซึ่งส่งผลต่อระบบประสาทส่วนกลาง เดิมเรียกว่า “โรคเดวิก” และเป็นเวลาหลายปีที่ถูกมองว่าเป็นชนิดย่อยของโรคปลอกประสาทเสื่อมแข็ง (MS ) อย่างไรก็ตาม หลังจากการค้นพบแอนติบอดีตนเองต่ออะควาพอริน-4 (AQP4-IgG) ในปี 2004 ก็ได้รับการยอมรับว่าเป็นโรคอิสระ
ภูมิหลังทางประวัติศาสตร์ : ในปี 1870 เซอร์โทมัส คลิฟฟอร์ด ออลบัตต์ บรรยายถึงความสัมพันธ์ระหว่างไขสันหลังอักเสบและความผิดปกติของเส้นประสาทตา เป็นครั้งแรก การวิจัยในภายหลังแสดงให้เห็นว่าเป็นโรคที่แตกต่างจาก MS และปัจจุบันเข้าใจภายใต้แนวคิดที่ครอบคลุมของ “NMOSD ”
ระบาดวิทยา มีดังนี้:
อุบัติการณ์ : อุบัติการณ์รายปีโดยประมาณของ AQP4+NMOSD คือ 0.4-7.3 ต่อล้านคน1) ความแตกต่างทางเพศ : อัตราส่วนเพศประมาณ 1:9 โดยเพศหญิงเด่นชัดอายุที่เริ่มเป็น : ส่วนใหญ่เกิดในวัยกลางคน (40–60 ปี) โดยมีจุดสูงสุดในช่วงปลายอายุ 30 ถึงต้นอายุ 40เชื้อชาติ : พบได้บ่อยในผู้ที่มีเชื้อสายแอฟริกันและเอเชีย1) ความสัมพันธ์กับการตั้งครรภ์ : ประมาณ 20–47% ของผู้หญิงมีอาการครั้งแรกระหว่างตั้งครรภ์หรือภายในหนึ่งปีหลังคลอดหรือแท้งบุตร
Q
NMOSD แตกต่างจากโรคปลอกประสาทเสื่อมแข็ง (MS) อย่างไร?
A
NMOSD เป็นโรคที่เกิดจากแอนติบอดีซึ่งกำหนดเป้าหมายไปที่ช่องน้ำ AQP4 บนแอสโตรไซต์ และแตกต่างโดยพื้นฐานจาก MS ในด้านพยาธิสรีรวิทยา การรักษา และการพยากรณ์โรค ใน NMOSD รอยโรคตามยาวที่ยาว (LETM) และโรคประสาทอักเสบแก้วนำแสงชนิดรุนแรงเป็นลักษณะเฉพาะ และข้อแตกต่างที่สำคัญอีกประการคือ ยาปรับเปลี่ยนโรคที่ได้ผลใน MS เช่น อินเตอร์เฟอรอนเบตา อาจกระตุ้นให้เกิดการกำเริบใน NMOSD
Chuai Y, et al. Paraneoplastic neuromyelitis optica spectrum disorder with dual AQP4-IgG and CRMP5 antibodies following thymectomy: a case report. Front Neurosci. 2026. Figure 1. PM
CI D: PMC13006573. License: CC BY.
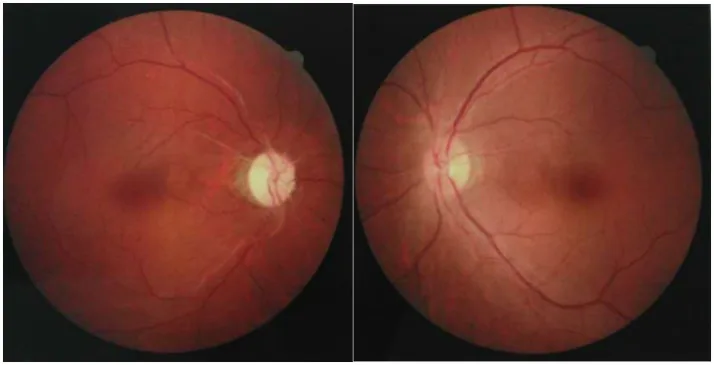
ภาพถ่าย
จอประสาทตา ของทั้งสองข้าง แสดงให้เห็นการ
ฝ่อของเส้นประสาทตา ที่มีหัวประสาทตาซีดในตาข้างหนึ่ง และการบวมเล็กน้อยของหัวประสาทตาในอีกข้างหนึ่ง ภาพเหล่านี้แสดงให้เห็นลักษณะของ
เส้นประสาทตา ในระยะเฉียบพลันถึงเรื้อรังของโรคประสาทอักเสบแก้วนำแสงใน
NMOSD อาการของ NMOSD มีความหลากหลายขึ้นอยู่กับบริเวณที่ได้รับผลกระทบ
การมองเห็น ลดลงอย่างเฉียบพลันสเตียรอยด์ ปวดตา ความผิดปกติของการมองเห็นสี ความบกพร่องของลานสายตา จุดบอดกลาง แต่รวมถึงตาบอดครึ่งซีกตามแนวนอน ตาบอดครึ่งซีกขมับทั้งสองข้าง และตาบอดครึ่งซีกแบบเดียวกัน เนื่องจากรอยโรคขยายไปถึงออปติกไคแอสมาออปติกแทรกต์ความผิดปกติทางประสาทสัมผัสและอัมพาตครึ่งล่าง : ความผิดปกติทางการเคลื่อนไหวและประสาทสัมผัสจากไขสันหลังอักเสบความผิดปกติของกระเพาะปัสสาวะและลำไส้ : ความผิดปกติของระบบประสาทอัตโนมัติที่เกิดร่วมกับไขสันหลังอักเสบอาการสะอึกที่รักษายาก คลื่นไส้ อาเจียน : อาการเฉพาะเนื่องจากรอยโรคที่ area postremaความผิดปกติของการเคลื่อนไหวลูกตา ภาวะนอนมากเกิน (คล้ายเฉียบหลับ) : เนื่องจากรอยโรคที่ไดเอนเซฟาลอน/ไฮโปทาลามัส
ในระยะเริ่มแรกอาจมีอาการคล้ายไข้หวัดใหญ่ (ไข้ ปวดกล้ามเนื้อ ปวดศีรษะ)
โรคประสาทตาอักเสบ
ภาวะบวมของจานประสาทตา : พบในระยะเฉียบพลัน ต่อมาจะพัฒนาเป็นฝ่อของประสาทตา
RAPD (ความบกพร่องของรูม่านตา ต่อแสงสัมพัทธ์)
เป็นพร้อมกันทั้งสองข้าง : โรคประสาทตาอักเสบ ใน NMOSD เป็นพร้อมกันทั้งสองข้างใน 17-82% ของผู้ป่วย แตกต่างจาก MS อย่างสำคัญ
การมองเห็น ลดลงอย่างรุนแรงNMOSD ค่ามัธยฐานของการมองเห็น ต่ำสุดอยู่ที่ระดับ hand motion (HM) หลังฟื้นตัว ค่ามัธยฐานยังคงอยู่ที่ระดับนับนิ้ว และ 60-69% มีความบกพร่องทางการมองเห็น ถาวรอย่างน้อย 20/200 ในตาข้างเดียว
ไขสันหลังอักเสบ
LETM (ไขสันหลังอักเสบตามขวางยาว) : รอยโรคต่อเนื่องยาว ≥3 กระดูกสันหลัง ประมาณ 85% ของ AQP4+NMOSD แสดงลักษณะนี้ในระยะไขสันหลังอักเสบเฉียบพลัน
กลุ่มอาการไขสันหลังสมบูรณ์ : เกี่ยวข้องกับทั้งสามทางเดิน คือ การเคลื่อนไหว ความรู้สึก และระบบประสาทอัตโนมัติ
ความบกพร่องทางการทำงานรุนแรง : ผู้ป่วยมากกว่า 30% ต้องใช้วีลแชร์ในช่วงที่อาการกำเริบรุนแรงที่สุด 37-44% ของ AQP4+NMOSD ในที่สุดต้องใช้อุปกรณ์ช่วยเดิน
กลุ่มอาการแอเรียโพสตรีมา (Area Postrema Syndrome)
อาการสะอึกที่รักษายาก : เกิดขึ้นต่อเนื่องเป็นเวลาหลายวันถึงหลายสัปดาห์ และไม่ตอบสนองต่อยาแก้อาเจียนทั่วไป
อาการคลื่นไส้และอาเจียน : เนื่องจากแอเรียโพสตรีมาไม่มีด่านกั้นเลือด-สมอง จึงเป็นตำแหน่งที่ AQP4-IgG สามารถเข้าถึงได้โดยตรง
ผลการตรวจวินิจฉัยหลักของ NMOSD : อาการสะอึกที่รักษายากโดยไม่มีสาเหตุชัดเจนเป็นสิ่งกระตุ้นให้สงสัย NMOSD อย่างจริงจัง
MS
โรคประสาทอักเสบแก้วนำแสงใน NMOSD รุนแรงกว่า กระจายเป็นบริเวณกว้างกว่า เป็นสองข้าง กลับเป็นซ้ำ และฟื้นตัวได้ยากกว่าเมื่อเทียบกับ MS การเกี่ยวข้องกับออปติกไคแอสมาคุณลักษณะเฉพาะของ NMOSD ใน MS จุดบอดกลาง ข้างเดียวเป็นลักษณะทั่วไป และพยากรณ์การมองเห็น ค่อนข้างดี
Q
โรคประสาทอักเสบแก้วนำแสงใน NMOSD แตกต่างจากที่สัมพันธ์กับ MS อย่างไร?
A
โรคประสาทอักเสบแก้วนำแสงใน NMOSD รุนแรงกว่า เป็นสองข้าง และกลับเป็นซ้ำ โดยพยากรณ์การมองเห็น ไม่ดี ใน NMOSD ที่มี AQP4 บวก ประมาณ 60-69% มีความบกพร่องทางการมองเห็น ถาวรอย่างน้อย 20/200 ในตาข้างเดียว นอกจากนี้ยังมีแนวโน้มเกี่ยวข้องกับออปติกไคแอสมา ทำให้เกิดความบกพร่องของลานสายตา ที่หลากหลาย เช่น ตาบอดครึ่งซีกขมับทั้งสองข้าง ซึ่งแตกต่างจาก MS
ยังไม่ทราบสาเหตุที่แน่ชัดของ NMOSD อย่างสมบูรณ์ เชื่อว่าการสูญเสียความทนทานต่อภูมิต้านตนเองเป็นสาเหตุพื้นฐาน
ปัจจัยเสี่ยงหลัก มีดังนี้:
เพศหญิง : อัตราส่วนเพศชายต่อหญิงประมาณ 1:9 โดยพบในเพศหญิงมากกว่าอย่างท่วมท้นเชื้อชาติ : ความเสี่ยงสูงขึ้นในประชากรเอเชียและแอฟริกาโรครูมาติกภูมิต้านตนเองร่วม : เช่น โรคลูปัส erythematosus ทั่วร่าง (SLE ), กลุ่มอาการโจเกรน, โรคกล้ามเนื้ออ่อนแรงชนิดร้ายแรง
10-30% ของผู้ป่วย NMOSD มีความสัมพันธ์กับกลุ่มอาการโจเกรน นอกจากนี้ยังมีรายงานในผู้ป่วยเด็ก 5)
การพบร่วมกับโรคกล้ามเนื้ออ่อนแรง (myasthenia gravis) พบได้ 2-3% ของผู้ป่วย1)
เนื้องอกมะเร็ง (NMOSD ชนิดพารานีโอพลาสติก) : ประมาณว่า 3-5% ของ NMOSD เป็นชนิดพารานีโอพลาสติก2) 3)
มีรายงานมะเร็งเต้านม มะเร็งปอด เทอราโทมาของรังไข่ และอื่นๆ3)
กลไกที่เสนอคือการแสดงออกของ AQP4 ในเนื้องอกกระตุ้นการตอบสนองทางภูมิคุ้มกันตนเอง2)
NMOSD ที่เกี่ยวข้องกับเทอราโทมาพบได้บ่อยในหญิงสาว (อายุเฉลี่ย 32.7 ปี)2)
หากคุณมีโรคภูมิต้านตนเอง เช่น กลุ่มอาการโจเกรนหรือ SLE และมีอาการตามัวเฉียบพลันหรือสะอึก/อาเจียนที่รักษายาก โปรดปรึกษาแพทย์ระบบประสาทหรือจักษุแพทย์ระบบประสาทโดยเร็ว โดยคำนึงถึงความเป็นไปได้ของ NMOSD
เกณฑ์การวินิจฉัย NMOSD ที่มี AQP4-IgG บวก คือการมีคุณสมบัติครบสามข้อต่อไปนี้:
มีลักษณะทางคลินิกหลักอย่างน้อยหนึ่งข้อ
AQP4-IgG บวก (โดยใช้วิธีตรวจที่ดีที่สุด)
ไม่พบโรคอื่นที่อธิบายอาการได้
เกณฑ์การวินิจฉัย NMOSD ที่มี AQP4-IgG ลบหรือไม่ได้ตรวจ คือการมีคุณสมบัติครบสี่ข้อต่อไปนี้:
มีลักษณะทางคลินิกหลักอย่างน้อยสองข้อ (หนึ่งในนั้นต้องเป็นโรคประสาทตาอักเสบ , LETM หรือกลุ่มอาการบริเวณ postrema)
การกระจายตัวในเชิงพื้นที่
เป็นไปตามข้อกำหนด MRI เพิ่มเติม
การแยกการวินิจฉัยอื่นออก
ลักษณะทางคลินิกหลัก (6 รายการ) มีดังนี้:
โรคประสาทอักเสบตา
ไขสันหลังอักเสบเฉียบพลัน
กลุ่มอาการบริเวณ postrema (สะอึกดื้อรักษา คลื่นไส้ อาเจียน)
กลุ่มอาการก้านสมองเฉียบพลัน
โรคลมหลับแบบมีอาการ/กลุ่มอาการไฮโปทาลามัสเฉียบพลัน
กลุ่มอาการสมองใหญ่แบบมีอาการ
ตารางต่อไปนี้แสดงการเปรียบเทียบวิธีการตรวจหาแอนติบอดีหลัก
วิธีการตรวจ ความไว ความจำเพาะ หมายเหตุ CBA (การทดสอบโดยใช้เซลล์เป็นฐาน) 69.7–100% 85.8–100% วิธีที่แนะนำ วิธี ELISA ด้อยกว่า CBA เล็กน้อย ด้อยกว่า CBA เล็กน้อย ครอบคลุมโดยประกันในญี่ปุ่น
AQP4-IgG : เฉพาะสำหรับ NMOSD แนะนำให้ตรวจวัดในช่วงที่มีอาการกำเริบเฉียบพลันและก่อนเริ่มการรักษาด้วยยากดภูมิคุ้มกัน1) CBA (การทดสอบโดยใช้เซลล์เป็นฐาน) : วิธีการตรวจที่แนะนำในปัจจุบัน อัตราผลบวกลวงของ ELISA รายงานว่าสูงกว่า CBA 5 เท่า1) MOG-IgG NMOSD ที่มีผล AQP4-IgG เป็นลบ1) แถบโอลิโกโคลนอลในน้ำไขสันหลัง (OCB) : ต่ำ (10–20%) ใน NMOSD (88% ใน MS ) ผลลบชี้แนะถึง NMOSD 1) จำนวนเม็ดเลือดขาวในน้ำไขสันหลัง : >50/μL การมีนิวโทรฟิลหรืออีโอซิโนฟิลช่วยแยก NMOSD ออกจาก MS ค่าการกะพริบวิกฤต (CFF ) : มีประโยชน์ในการประเมินกิจกรรมของโรคประสาทอักเสบแก้วนำแสง ลดลงใน NMOSD
MRI ไขสันหลัง : LETM เป็นลักษณะที่เด่นชัดที่สุด. เน้นที่เนื้อเทาส่วนกลาง. ร่วมกับไขสันหลังบวม, สัญญาณต่ำใน T1, และการเพิ่มความเข้มของ Gd. ประมาณ 85% ของผู้ป่วย NMOSD ที่มี AQP4+ แสดง LETM ในระหว่างไขสันหลังอักเสบเฉียบพลัน 1) MRI เส้นประสาทตา : ลำดับการระงับสัญญาณไขมันเป็นสิ่งจำเป็น. การอักเสบทั้งสองข้างและยาว (>50%) เป็นลักษณะเฉพาะ. การเกี่ยวข้องกับส่วนหลังและออปติกไคแอสมาพบได้บ่อยใน AQP4+ NMOSD 1) MRI สมอง : รอยโรคบริเวณ area postrema, รอยโรคก้านสมองรอบช่องที่สี่, รอยโรคไฮโปทาลามัส/รอบช่องที่สาม, และรอยโรคเนื้อขาวกระจาย.ลักษณะของ NMOSD : แตกต่างจาก MS , รอยโรค T2 ใหม่ที่ไม่มีอาการพบได้น้อย (3-13%). โดยทั่วไปไม่จำเป็นต้องทำ MRI เฝ้าระวัง 1)
โรคปลอกประสาทเสื่อมแข็ง (MS )
โรคที่เกี่ยวข้องกับแอนติบอดี MOG (MOGAD )โรคสมองและไขสันหลังอักเสบเฉียบพลันแพร่กระจาย (ADEM )
โรคลูปัส erythematosus ทั่วร่าง
โรคเบห์เซ็ททางระบบประสาท
ในผู้สูงอายุ สิ่งสำคัญคือต้องแยกจากโรคเส้นประสาทตา ขาดเลือด, โรคไขสันหลังส่วนคอ, ไขสันหลังขาดเลือด, และมะเร็งต่อมน้ำเหลืองระบบประสาทส่วนกลางปฐมภูมิ.
Q
NMOSD สามารถวินิจฉัยได้แม้ว่าแอนติบอดี AQP4 จะเป็นลบหรือไม่?
A
ได้. แม้ว่า AQP4-IgG จะเป็นลบหรือไม่ได้ตรวจสอบ หากมีลักษณะทางคลินิกหลักสองข้อขึ้นไป, ตรงตามข้อกำหนด MRI เพิ่มเติม, และไม่รวมโรคอื่นๆ ก็สามารถวินิจฉัย NMOSD ได้. นอกจากนี้ ประมาณ 30% ของกรณี AQP4-IgG ลบมี MOG-IgG บวก, และแนะนำให้วัดแอนติบอดีทั้งสองชนิด. น้อยกว่า 1% ของกรณี AQP4-IgG ลบจะเปลี่ยนเป็นบวกในภายหลัง.
ทางเลือกแรก: การรักษาด้วยสเตียรอยด์แบบพัลส์
เมทิลเพรดนิโซโลน 1,000 มก./วัน ฉีดเข้าหลอดเลือดดำเป็นเวลา 3 วัน
หากการมองเห็น ไม่ดีขึ้น ให้พิจารณาทำซ้ำอีก 1 คอร์สหลังจากเว้นช่วง 3-4 วัน
โรคประสาทอักเสบแก้วนำแสงใน NMOSD มีการดื้อต่อสเตียรอยด์ สูง ดังนั้นหากตอบสนองไม่เพียงพอ ควรพิจารณาการรักษาต่อไปตั้งแต่เนิ่นๆ
ทางเลือกที่สอง: การรักษาด้วยการแลกเปลี่ยนพลาสมา
ดำเนินการเมื่อไม่ตอบสนองต่อสเตียรอยด์ พัลส์ ตัวเลือกต่อไปนี้มีให้:
การแลกเปลี่ยนพลาสมา แบบธรรมดา (PE)การแลกเปลี่ยนพลาสมา แบบกรองสองชั้น (DFPP)การบำบัดด้วยการดูดซับภูมิคุ้มกัน (IA) : สามารถกำจัดแอนติบอดีแบบเลือกสรรได้
ลำดับประสิทธิภาพถือว่า การแลกเปลี่ยนพลาสมา แบบธรรมดา > การกรองสองชั้น > การดูดซับภูมิคุ้มกัน ดำเนินการ 5-6 ครั้งต่อคอร์ส และหลังการรักษาต้องนอนโรงพยาบาลจนกว่าระดับ IgG ในร่างกายจะฟื้นตัว หมายเหตุ: สำหรับ “โรคประสาทอักเสบแก้วนำแสง” อาจไม่ครอบคลุมโดยประกัน จึงต้องอธิบายให้ผู้ป่วยทราบ
ใน AQP4+NMOSD ควรเริ่มการรักษาแบบประคับประคองตั้งแต่เนิ่นๆ หลังการโจมตีครั้งแรก 1) หลังการแลกเปลี่ยนพลาสมา มักเปลี่ยนไปใช้เพรดนิโซโลน 5-10 มก./วัน + อะซาไธโอพรีน 50-100 มก./วัน
สารชีวภาพที่มีระดับหลักฐานสูง มีดังนี้:
ยาที่ยับยั้งคอมพลีเมนต์
อีคูลิซูแมบ : 900 มก. IV ทุกสัปดาห์ × 4 ครั้ง → 1,200 มก. ทุก 2 สัปดาห์เป็นขนาดประคับประคอง 1) ราวูลิซูแมบ (Ravulizumab) : ขนาดยาเริ่มต้นตามน้ำหนักตัว (2,400–3,000 มก.) → หลังจากวันที่ 15 ให้ 3,000–3,600 มก. ทุก 8 สัปดาห์1)
การบำบัดด้วยการกำจัดเซลล์บี
ริทูซิแมบ (Rituximab)1) อินิบิลิซูแมบ (Inebilizumab) : 300 มก. ทางหลอดเลือดดำ ทุก 15 วัน จำนวน 2 ครั้ง → ทุก 6 เดือน1)
ยาที่ยับยั้งตัวรับ IL-6
ซาทราลิซูแมบ (Satralizumab) : 120 มก. ฉีดใต้ผิวหนัง ทุก 4 สัปดาห์1)
อินเตอร์เฟียรอนเบตา นาทาลิซูแมบ และฟิงโกลิโมดที่ใช้ในโรค MS อาจกระตุ้นหรือทำให้อาการกำเริบของ NMOSD แย่ลง จึงควรถือเป็นข้อห้ามใช้
ยาป้องกันการกำเริบต้องใช้ต่อเนื่องในระยะยาว และไม่ควรหยุดยาโดยไม่ปรึกษาแพทย์
การรักษาด้วยการแลกเปลี่ยนพลาสมา สำหรับโรคประสาทอักเสบแก้วนำแสงอาจไม่ครอบคลุมโดยประกัน ดังนั้นควรตรวจสอบกับสถานพยาบาลล่วงหน้า
Q
จะทำอย่างไรหากการรักษาด้วยสเตียรอยด์ขนาดสูงไม่ได้ผล?
A
การแลกเปลี่ยนพลาสมา เป็นทางเลือกถัดไป มีสามประเภท: การแลกเปลี่ยนพลาสมา อย่างง่าย, การแลกเปลี่ยนพลาสมา ด้วยการกรองเมมเบรนคู่, และการดูดซับภูมิคุ้มกัน การแลกเปลี่ยนพลาสมา อย่างง่ายถือว่ามีประสิทธิภาพสูงสุด แต่ก็สร้างภาระต่อร่างกายมากที่สุด หนึ่งคอร์สประกอบด้วย 5–6 ครั้ง และหลังการรักษาต้องเข้ารับการรักษาในโรงพยาบาล
NMOSD โดยพื้นฐานแล้วเป็นโรคของแอสโทรไซต์ (astrocytopathy) กลไกการเกิดโรคมีดังนี้:
การผลิตแอนติบอดีและการผ่านด่านกั้นเลือด-สมอง (BBB)
ในส่วนปลาย เซลล์บีจะแยกตัวเป็นพลาสมาบลาสต์ที่หลั่ง AQP4-IgG IL-6 ช่วยส่งเสริมการแยกตัวนี้และเพิ่มการซึมผ่านของเลือด-สมอง barrier บริเวณ area postrema เป็นบริเวณที่ไม่มี blood-brain barrier และอาจเป็นเส้นทางที่ AQP4-IgG เข้าสู่ระบบประสาทส่วนกลาง
สายโซ่ของการทำลายแอสโตรไซต์
AQP4-IgG จับกับช่องน้ำ AQP4 ที่แสดงออกอย่างหนาแน่นบนเท้าแอสโตรไซต์ ทำให้เกิดการบาดเจ็บของแอสโตรไซต์ผ่านวิถีทางดังต่อไปนี้
การกระตุ้นวิถีคลาสสิกของคอมพลีเมนต์ : ส่วน Fc ของ AQP4-IgG กระตุ้นคอมพลีเมนต์ เกิดเป็น membrane attack complex (MAC) ซึ่งทำลายแอสโตรไซต์โดยตรงADCC (antibody-dependent cell-mediated cytotoxicity) : เซลล์ NK และนิวโทรฟิลทำลายแอสโตรไซต์ผ่าน Fcγ receptorการปล่อย C5a anaphylatoxin : ดึงดูดแกรนูโลไซต์ (นิวโทรฟิล, อีโอซิโนฟิล) ทำให้เกิดความเสียหายต่อแอกซอนทุติยภูมิและการทำลายไมอีลิน1)
ความแตกต่างทางพยาธิวิทยาจาก MS
ใน MS การทำลายไมอีลินที่เนื้อขาวเป็นหลักโดยเซลล์ T CD8+ เป็นหลัก ในขณะที่ NMOSD เซลล์ T CD4+ มีส่วนร่วมมากกว่า ทำให้เกิดรอยโรคเนื้อตายที่เกี่ยวข้องทั้งเนื้อเทาและเนื้อขาว
เหตุผลของการกระจายตัว
ช่อง AQP4 กระจายตัวมากในเส้นประสาทตา , area postrema และไขสันหลัง ทำให้บริเวณเหล่านี้เป็นเป้าหมายเฉพาะ
ตัวบ่งชี้ทางชีวภาพ
GFA P ในซีรัม : สะท้อนการบาดเจ็บของแอสโตรไซต์และมีค่าสูงในช่วงที่มีอาการกำเริบNeurofilament light chain (NfL) ในซีรัม : สะท้อนความเสียหายของแอกซอนและสัมพันธ์กับความรุนแรงของอาการกำเริบ1)
ไซโตไคน์ที่เกี่ยวข้อง ได้แก่ IL-6, IL-10, IL-17a, G-CSF, TNF -α และ BAFF/APRIL ได้รับการรายงานแล้ว
ประมาณว่า 3-5% ของ NMOSD เป็นแบบพารานีโอพลาสติก กรณีที่เกี่ยวข้องกับเทราโทมาของรังไข่ได้รับการศึกษาอย่างละเอียด
Ikeguchi และคณะ (2021) ได้ทบทวน 6 กรณีของ NMOSD ที่เกี่ยวข้องกับเทราโทมาของรังไข่และมี AQP4 บวก2) ผู้ป่วยทั้งหมดเป็นผู้หญิง อายุเริ่มป่วยเฉลี่ย 32.7 ปี (15-50 ปี) ใน 6 กรณี 83% (5/6) มีอาการคลื่นไส้อาเจียน 83% มีแถบโอลิโกโคลนอลในน้ำไขสันหลังเป็นบวก และ 83% มีรอยโรคที่ก้านสมองส่วนหลัง การวิเคราะห์ทางพยาธิวิทยายืนยันปฏิกิริยาทางภูมิคุ้มกันของ AQP4 และการแทรกซึมของลิมโฟไซต์ในเนื้อเยื่อประสาทที่ติด GFA P บวกภายในเนื้องอก ชี้ให้เห็นกลไกที่การนำเสนอแอนติเจน AQP4 ภายในเนื้องอกกระตุ้นการตอบสนองของภูมิต้านตนเอง หลังการตัดเนื้องอก AQP4-IgG กลายเป็นลบใน 60% (3/5) ของกรณี
Ding และคณะ (2021) ได้ทบทวน 43 กรณีของ NMOSD แบบพารานีโอพลาสติก3) 88.4% เป็นผู้หญิง โดยมะเร็งเต้านมและมะเร็งปอดเป็นชนิดเนื้องอกที่พบบ่อยที่สุด มีการเน้นย้ำถึงความสำคัญของการตรวจคัดกรองเนื้องอก โดยเฉพาะในผู้ป่วย NMOSD ที่อายุมากกว่า 50 ปี
แนะนำให้ตรวจคัดกรองเนื้องอก รวมถึงเทราโทมา แม้ในกรณีผู้ป่วยอายุน้อย
มีการรายงานประสิทธิภาพของการบำบัดด้วยการดูดซับภูมิคุ้มกันโปรตีนเอ (IA) ใน NMOSD ที่ดื้อต่อการรักษาซึ่งไม่ตอบสนองต่อสเตียรอยด์ การแลกเปลี่ยนพลาสมา หรือริตูซิแมบ
Fan และคณะ (2024) ได้ทำการดูดซับภูมิคุ้มกันโปรตีนเอ 3 ครั้งในหญิงอายุ 35 ปีที่มี NMOSD ร่วมกับกลุ่มอาการโจเกรนชนิดดื้อต่อการรักษาซึ่งไม่ตอบสนองต่อการให้สเตียรอยด์ แบบพัลส์และ IVI G4) ภายในหนึ่งสัปดาห์ ความผิดปกติทางการมองเห็น อัมพาตครึ่งล่าง และความผิดปกติของการรับรู้ตำแหน่งดีขึ้นอย่างมีนัยสำคัญ และพบว่ามีการลดลงอย่างรวดเร็วของ AQP4-IgG, IgA, IgG และ IgM ไม่พบการกลับเป็นซ้ำหรือการดำเนินโรคในช่วง 4 ปีของการติดตามผล
ภาพที่แท้จริงของโรค autoimmune ต่างๆ ที่เกิดร่วมกับ NMOSD กำลังเริ่มชัดเจนขึ้น
Zhu และคณะ (2025) รายงานกรณีของเด็กหญิงอายุ 14 ปีที่เริ่มป่วยเป็น NMOSD เมื่ออายุ 11 ปี5) ผล AQP4-IgG เป็นบวก และได้รับการยืนยันว่าเป็นกลุ่มอาการโจเกรนปฐมภูมิในระหว่างการดำเนินโรค การรักษาให้อยู่ในระยะสงบด้วย methylprednisolone, IVI G และ mycophenolate mofetil (MMF) จากนั้นเปลี่ยนเป็น tacrolimus มีรายงานโรคร่วม autoimmune ใน 20-30% ของกรณี NMOSD ในผู้ใหญ่ แต่กรณีนี้แสดงให้เห็นว่าโรคร่วมนี้สามารถเกิดขึ้นได้ในเด็กเช่นกัน5)
Cacciaguerra L, Flanagan EP. Updates in NMOSD and MOGAD Diagnosis and Treatment. Neurol Clin. 2024;42(1):77-114.
Ikeguchi R, Shimizu Y, Shimomura A, Suzuki M, Shimoji K, Motohashi T, Yamamoto T, Shibata N, Kitagawa K. Paraneoplastic AQP4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorder Associated With Teratoma: A Case Report and Literature Review. Neurol Neuroimmunol Neuroinflamm. 2021;8(5):e1045. doi:10.1212/NXI.0000000000001045. PMID: 34285095. PMCI D: PMC8293286.
Ding M, Lang Y, Cui L. AQP4-IgG positive paraneoplastic NMOSD : A case report and review. Brain and behavior. 2021;11(10):e2282. doi:10.1002/brb3.2282. PMID:34520629; PMCI D:PMC8553315.
Fan W, Chen X, Xiao P, Wei B, Zhang Y, Huang J, et al. Protein-A immunoadsorption combined with immunosuppressive treatment in refractory primary Sjögren’s syndrome coexisting with NMOSD : a case report and literature review. Frontiers in immunology. 2024;15:1429405. doi:10.3389/fimmu.2024.1429405. PMID:39055718; PMCI D:PMC11269126.
Zhu G-q, Hu R-x, Peng Y, et al. A Chinese girl with neuromyelitis optica spectrum disorder coexisting with primary Sjogren’s syndrome: a case report and literature review. Front Immunol. 2025;16:1559825.